- Sturge᠆Weber 症候群
病 態:
皮膚・眼・髄膜(脳軟膜)に発症する(静脈性)血管腫を特徴とする過誤腫 hamartoma(血管奇形)で,古典的には,顔面の port᠆wine母斑,緑内障(牛眼),脳内の軟膜血管腫を有する病態である.
皮膚病変は眼瞼~三叉神経(主に第一枝,第二枝)の支配領域に出現する血管腫 nevus flammeusである.
眼圧上昇は 48~71% にみられ,早期ではおもに隅角の形成異常・Schlemm管萎縮により,晩期では上強膜血管腫に伴う上強膜静脈圧の上昇や周辺虹彩前癒着の形成による.脈絡膜血管腫はびまん型でしばしば続発網膜剝離を発症する.
顔面血管腫と同側に髄膜血管腫や頭蓋内石灰化を伴うことがある.これらにより,難治性てんかん,精神発達遅滞,運動麻痺などが問題となる.
発症は(母斑症のカテゴリーとして)胎生期の交感神経の障害による血管(静脈)形成異常が病因と考えられ,最近GNAQ遺伝子の体細胞モザイク変異が報告された.先天性疾患であるが,一般的に遺伝性はないとされる.
責任遺伝子:
GNAQ(G protein subunit alpha q遺伝子;9q21.2 ,別名SWS,など.
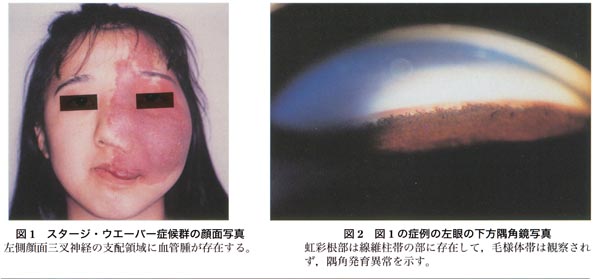 |
| (引用:臨眼 53(5), 1999 |
治 療:
緑内障は原因が複合していると考えられ,かつ,診断時が年少であることで対処が難しい.さらに脈絡膜血管腫についても決め手がない.
☑ Roach Scale
type Ⅰ: 顔面病変,脈絡叢病変,頭蓋内病変 pial angioma,および緑内障が揃う場合(classic form).
type Ⅱ: 頭蓋内病変を伴わない顔面病変のみ.緑内障が伴うこともある.
type Ⅲ: 顔面病変を伴わない頭蓋内病変 pial angiomaのみの場合.通常,緑内障は伴わない.
☑ 片側性,両側性ともに生じる.本症の 95%以上で皮膚病変をみる.
☑ port-wine stain(皮膚の単純性血管腫):Sturge᠆Weber症候群(SWS)では必ず顔面三叉神経第一枝を含む.ただし発生上の発症基盤は血管腫と三叉神経とは関連がなく偶然の共通範囲,とのことである.なお,第二枝・三枝のみのときはSWSではない.
- von Hippel᠆Lindau (retinal capillary hemangioblastoma)
☞
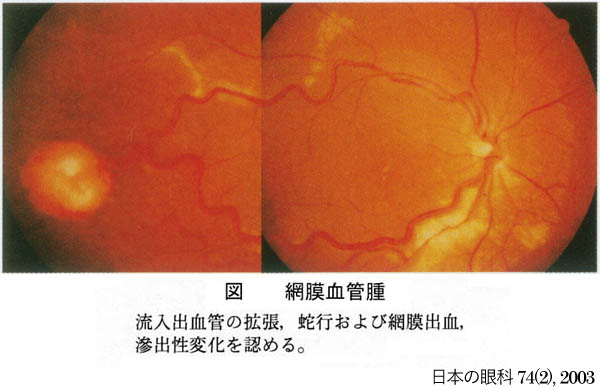
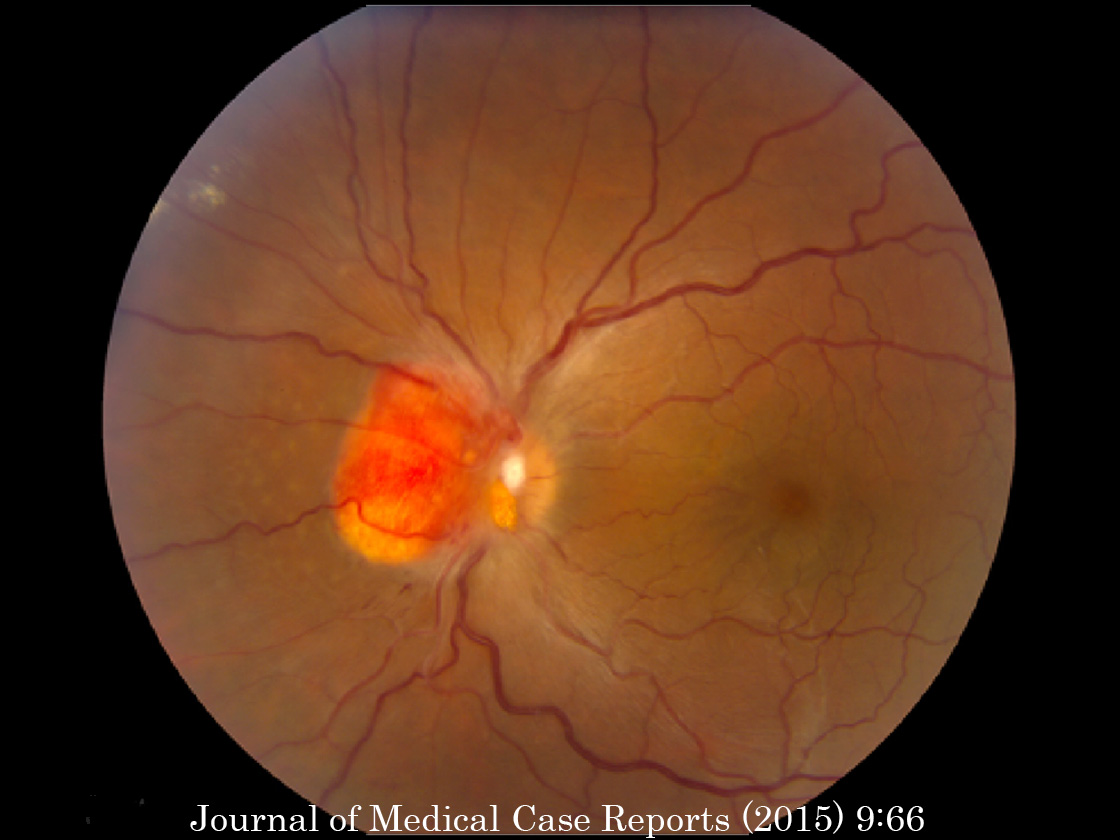
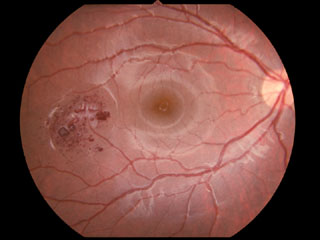
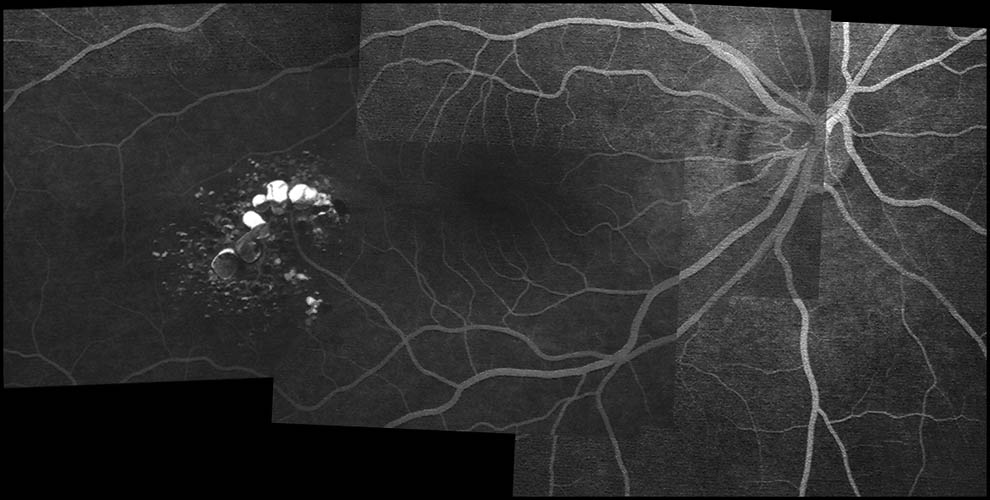
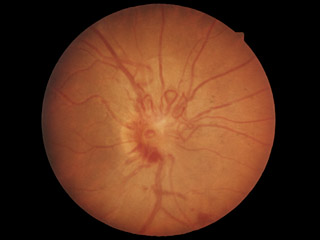
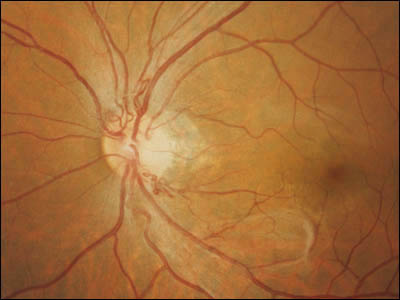
症例写真 ➂
,
➃
病 態:
脳,内耳,眼などの中枢神経系と腎,副腎,膵などの内臓に多発性の出血性腫瘍(良性/悪性)をきたす.初め von Hippel による網膜血管芽腫が報告され,続いて
Lindau
が内臓の嚢腫変化を伴う小脳と網膜の血管芽腫を記載し,本疾患名となっている.罹患者の60%に眼所見があり,1∕3~半数が両眼性.だいたいが周辺部の血管腫のため,滲出病変が黄斑に届いて視力障害を発症することになる.15%ほどで,傍乳頭血管腫が生じる.
分 類:
①第1期:動静脈拡張,異常蛇行,血管芽腫形成
②第2期:網膜出血,滲出性病変
③第3期:著明な滲出性病変,網膜剝離
④第4期:網膜・隣接組織の破壊,ぶどう膜炎,血管新生緑内障
責任遺伝子:
VHL(von Hippel−Lindau tumor suppressor遺伝子;3p25.3 ,腫瘍抑制遺伝子の変異.低酸素誘導因子(hypoxia−inducible factor:HIF)の不活化がうまくゆかず,血管内皮細胞増殖因子(vascular endothelial growth factor:VEGF)や血小板由来増殖因子(platelet−derived growth factor:PDGF)などの制御に失敗することが基本変化,と考えられている.
常染色体優性遺伝形式.
- 神経線維腫症 neurofibromatosis (von Recklinghausen syndrome)
病 態:
neurofibromatosis(NF)は,皮膚の異常色素沈着と神経外胚葉系にできる腫瘍を特徴とし,いくつかのサブタイプがある(NF-1~NF-7).代表的には,neurofibromatosis type1 と type2 の2つ.
両者はまったく別の疾患で,それぞれ異なった染色体上の遺伝子に起因する.

症状・分類:
眼科領域では網膜・虹彩・眼瞼の線維腫がみられ,両眼性あるいは片眼性の眼球突出を呈する(搏動性の時がある).
❉ type Ⅰ:NF1(peripheral neurofibromatosis,von Recklinghausen病)
皮膚の多発性の神経線維腫,いわゆる café au lait斑が特徴.虹彩の結節(色素性過誤腫;Lisch結節)は生下時には不明瞭であるが,成人では100%.網膜・脈絡膜の過誤腫のほか視神経膠腫を発症.房水排出路の腫瘍では緑内障を発症.
【
Lisch
】
そのほか学習障害,macrocephaly,bony abnormalities and malignancy など が乳幼児期に発症.
責任遺伝子:NF1(neurofibromin 1遺伝子;17q11.2 ,常染色体優性遺伝.
❉ type Ⅱ:NF2(central neurofibromatosis,bilateral acoustic neurofibromatosis)
通常10代以降で発症,両側性の vestibular schwannomas あるいは acoustic neuromas(聴神経腫
責任遺伝子:NF2(neurofibromin 2遺伝子;22q12.2 ,常染色体優性遺伝.シュワノミン遺伝子.
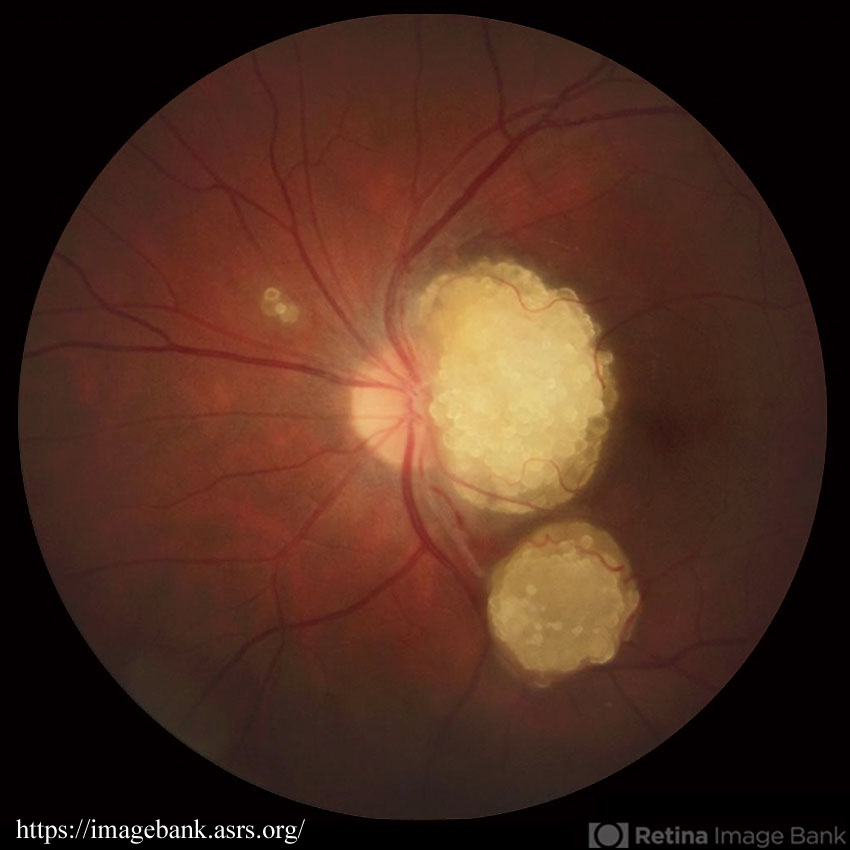
- 結節性硬化症 tuberous sclerosis (Bourneville᠆Pringle disease)
病 態:
Bourneville−Pringle
病は,過誤腫が,脳,皮膚,内臓,眼に複合(tuberous sclerosis complex)して出現する.
症状・分類:
皮膚の脱色素(葉状白斑),顔面の血管線維腫,脳室壁に膨隆した結節(石灰化),再発性てんかん発作・精神発達遅滞,網膜過誤腫を特徴とする.網膜あるいは視神経乳頭部に認められる眼底病変は astrocyte の過誤腫(神経膠腫:しばしば桑実様病変と形容)である.
【
皮脂腺腫
】
責任遺伝子:
TSC1(tuberous sclerosis 1遺伝子;9q34.13),TSC2(tuberous sclerosis 2遺伝子;16p13.3),における制御遺伝子(細胞の増殖や細胞の大きさの制御)の機能不全による常染色体優性(顕性)遺伝遺伝.繊毛病(ciliopathy).60%近くが孤発例といわれる.
- 太田母斑 nævus Ota (眼上顎部褐青色母斑 nævus fuscocæruleus ophthalmomaxillaris)
病 態:
真皮メラノサイトのメラニン産生 と 表皮メラノサイトのメラニン産生過剰による.
症状・分類:
片側の,三叉神経第一枝または第二枝に生じるmelanocyte(neural crest由来)による色素性母斑.眼科領域では黒褐色~青色の眼瞼縁,虹彩の多色素,結膜の斑状色素,強膜の青黒色斑等.眼底ではぶどう膜の青黒色斑のため,全体が暗くみえる.
責任遺伝子:一般に遺伝性はない.
- 色素失調症 incontinentia pigmenti (Bloch−Sulzberger 症候群)
病 態:
メラノブラストの一次的先天性機能障害.
症状・分類:
生下時から,紅斑を伴う水疱(炎症期)→丘疹(苔癬期)→色素沈着(色素沈着期)→消退(色素沈着消退期)という特徴的な経過の皮疹を生じる.皮膚のほか眼,毛髪,歯,中枢神経を障害する.
眼症状の中心は網膜毛細血管の発育障害で,網膜血管閉塞は生後1年以内に生じ,その後ほぼ停止する.高度であると滲出性網膜硝子体症あるいは未熟児網膜症類似の増殖網膜症により,6歳までに網膜剝離を発症する 牽引乳頭グループ の一つ.他にも多様な眼所見(斜視,先天白内障,視神経異常,黄斑低形成など)の報告がある.免疫不全を伴い,易感染性を示す(外胚葉形成不全免疫不全症.
責任遺伝子:
NEMO(NFⲻκB nuclear factor essential modulator)遺伝子(IKBKG遺伝子;Xp28)の変異による外胚葉形成不全.X連鎖優性遺伝形式.
- von Hippel᠆Lindau 病 (retinal capillary hemangioma
☝
母斑症 で
病 態:
3番染色体(3p25)VHL遺伝子の異常によりVEGFの過剰発現をきたす.これにより両眼性で常染色体優性遺伝を示す.
➊von Hippel病:網膜血管(芽)腫
から網膜実質・網膜下腔への漿液漏出により,進行性の網膜浮腫・続発性網膜剝離をきたす.
➋von Hippel᠆Lindau病:
小脳血管腫を伴う病態で,てんかん・精神障害・痴呆などを伴うことが多い.
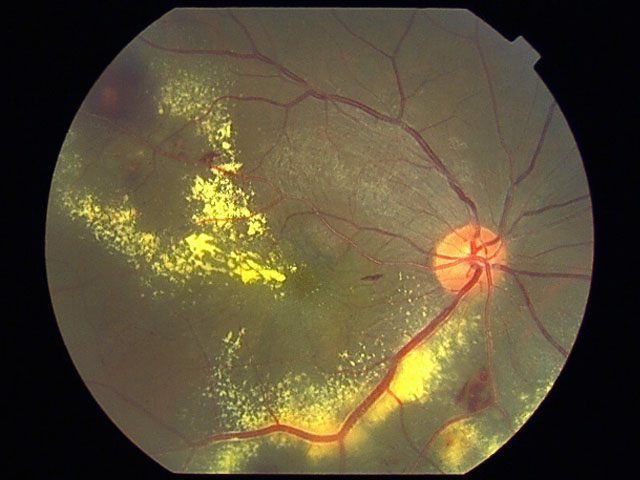
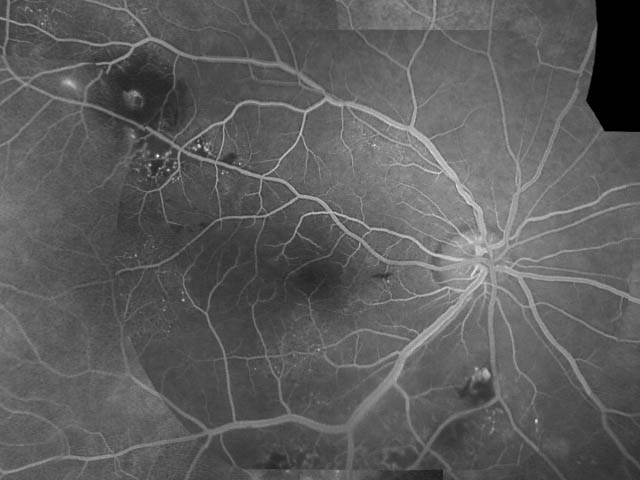
- Leber᠆Coats
病 態:
網膜血管内皮の機能的・構造的異常のため主に毛細血管に血管拡張・血管瘤形成,透過性亢進から滲出性病変を起こす.滲出が旺盛なまま進行すると,網膜剝離(滲出性網膜剝離)の状態となる.
網膜内滲出でとどまると Leber型,続発剝離をきたすと Coats型となる.
遺伝的な責任部位には,3番染色体,13番染色体(13q 12.1),X染色体(NDP gene)が報告されている.
遺伝性はないとされ,通常片眼性である.
分類・所見:
➊Leber病:(miliary angiomatosis:粟粒血管腫症)
進行が緩やかであることから,中年以降に視力障害を自覚する.一見,網膜細動脈瘤そっくりに発症することもあるが,長期経過のうちに多発性の血管瘤に悩むことになる.造影検査では毛細血管瘤以外にも透過性亢進がみられ,治療範囲の決定に苦慮する.
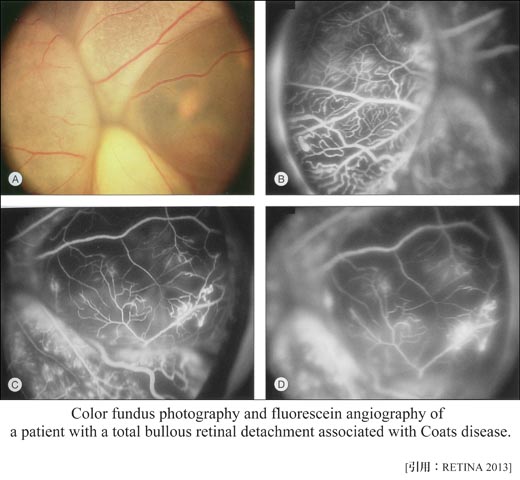
➋Coats病:(exudative retinitis:滲出性網膜炎)
小児(4∕5が男児)であり経過が長いため,浮腫液による硬性白斑(濃縮したリピッド,コレステリン結晶)の沈着や網膜剝離を起こしており,強い視力障害を生じていることが多く,さらに網膜芽細胞腫との鑑別が必要になる(白色瞳孔の原因の 一).
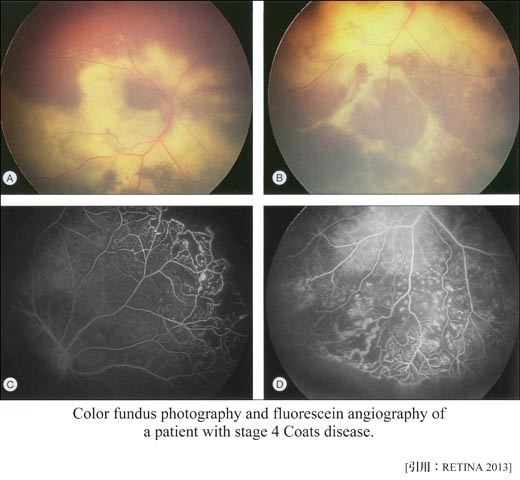
☑stage分類 (Am J Ophthalmol 2001)
| stage | 所見 | 割合 |
|---|
| 1 | 毛細血管の拡張 | 1% |
| 2 | 滲出変化が加わる | 14% |
| 2A | | 黄斑外 | |
| 2B | | 黄 斑 | |
| 3 | 滲出性網膜剝離の発症 | 69% |
| 3A1 | | 黄斑外の部分剝離 | |
| 3A2 | | 黄斑を含む部分剝離 | |
| 3B | | 全剝離 | |
| 4 | 全剝離と緑内障発症 | 15% |
| 5 | 眼球癆 | 2% |
| | |
☑exudative retinitis or exudative retinopathy?
治 療:
1. 方針
透過性の亢進した異常血管を凝固破壊することが治療の基本となる.遠慮して治療範囲を小さく設定すると治らなかったりする.
2. 実際
年少児に対しては全身麻酔下で,双眼倒像鏡に組み込んだアルゴンレーザーによる凝固を行うが,網膜冷凍凝固を行う場合もある.年長児〜成人では,通常の細隙灯顕微鏡下でアルゴンレーザーによる光凝固を行う.
3.これから
重症例では抗VEGF薬の適応が試みられる.
- 傍中心窩毛細血管拡張 parafoveal(juxtafoveolar ‥とも)telangiectasia
☞
症例写真
病 態:
おもに中心窩周囲毛細血管係蹄の耳側側に,血管拡張と透過性亢進をきたすもの.
他疾患特に,糖尿病網膜症・網膜静脈閉塞症が否定される必要がある.
症 状:
透過性亢進による黄斑浮腫を来たして視力が低下する.
分 類:
先天性・後天性,片眼性・両眼性,などで区別する(現実に可能か,意味があるかは ? ).
| Yannuzzi による分類(2006) |
| group 1 | aneurysmal | (血管瘤型) |
| group 2 | perifoveal | (血管拡張型) |
| group 3 | occlusive | (血管閉塞型) |
先天性は,Leber᠆Coats病のスペクトラム上にある(亜型)と理解されている.
治 療:
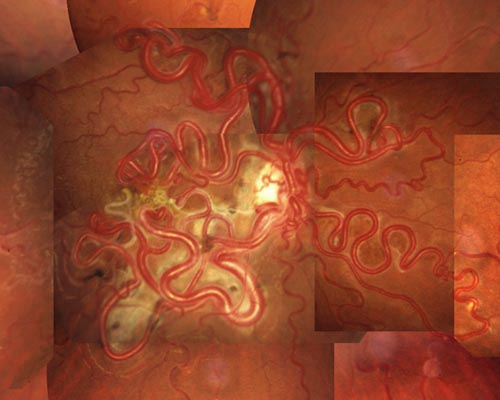
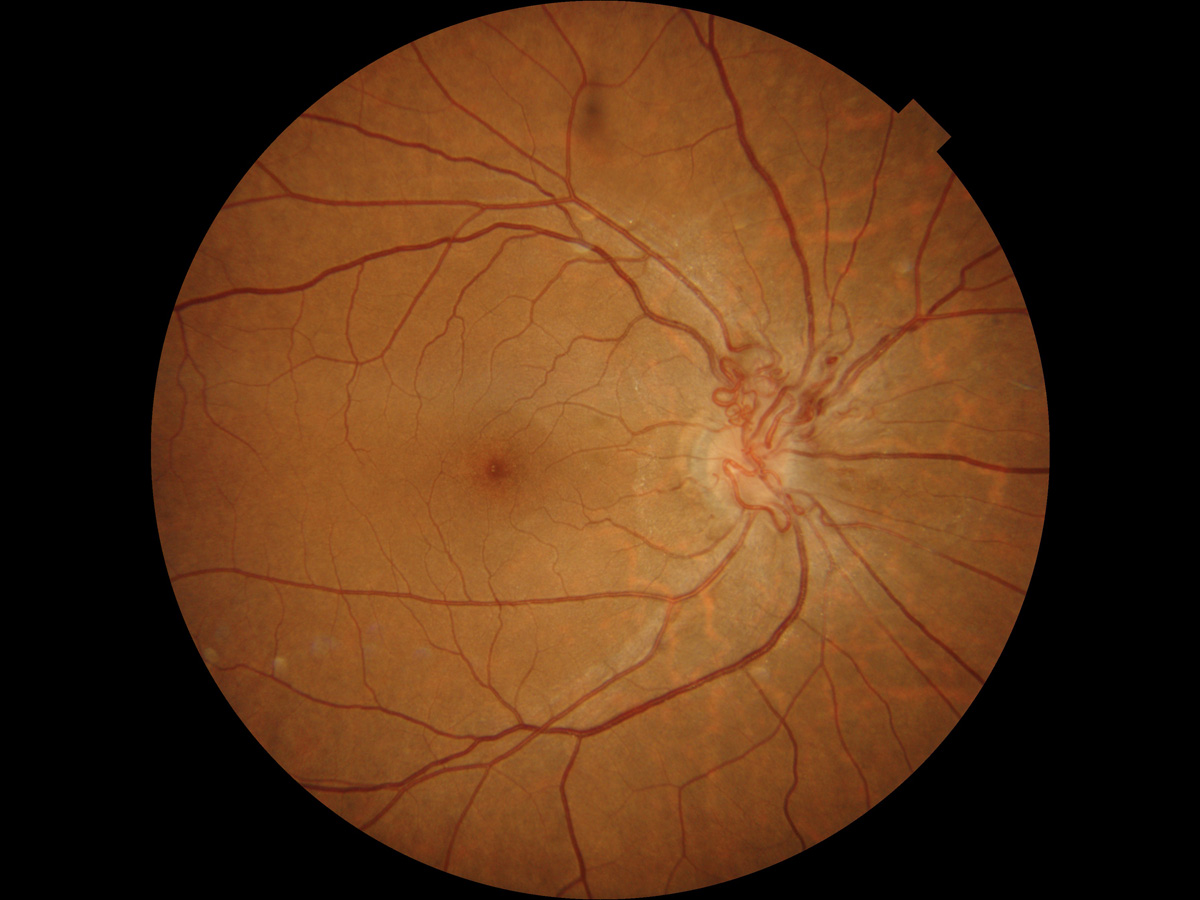
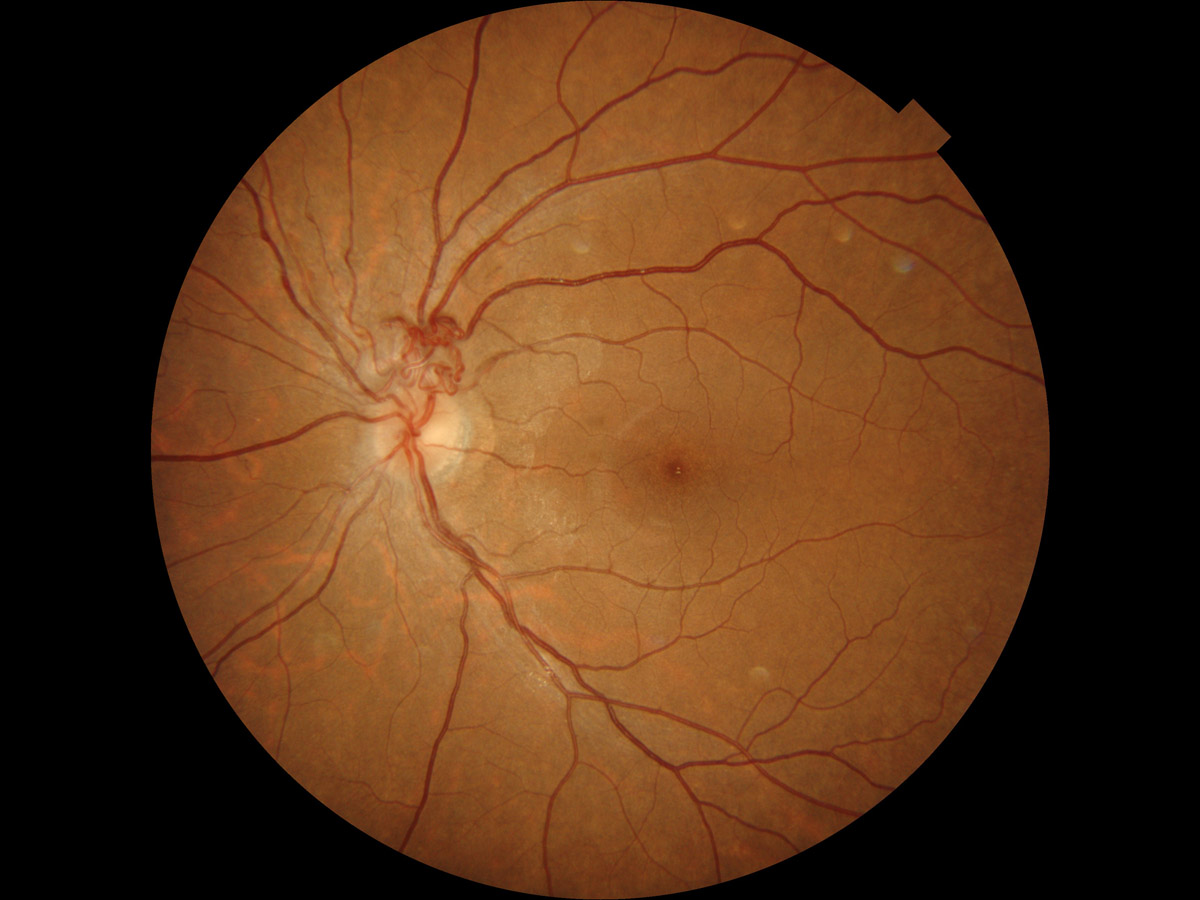
- 網膜細動脈瘤 retinal arteriolar macroaneurysm
☞
症例写真① , 症例写真②
病 態:
高血圧や動脈硬化のある高齢者に多く,第三分枝以内に発生する.透過性亢進を伴う.血栓が出来るようになると,破綻性にあるいは動脈瘤破裂として出血する.
症 状:
出血や滲出性変化が中心窩に及ばない限り,視力予後は悪くない(要するに,無自覚・無症状).
分 類:
血管腫のカテゴリーというコンセンサスはないが,Leber病に類似して細動脈瘤が次々に出来る case にも遭遇する.
治 療:
- capillary hemangioma on the optic disc(視神経乳頭部血管腫;vasoproliferative tumors of the ocular fundus
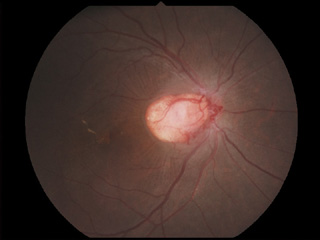
- cavernous hemangioma of the retina(海綿状血管腫
- retinal arteriovenous malformation
racemose hemangioma(蔓状血管腫
- racemose hemangioma combined with venous loop formation
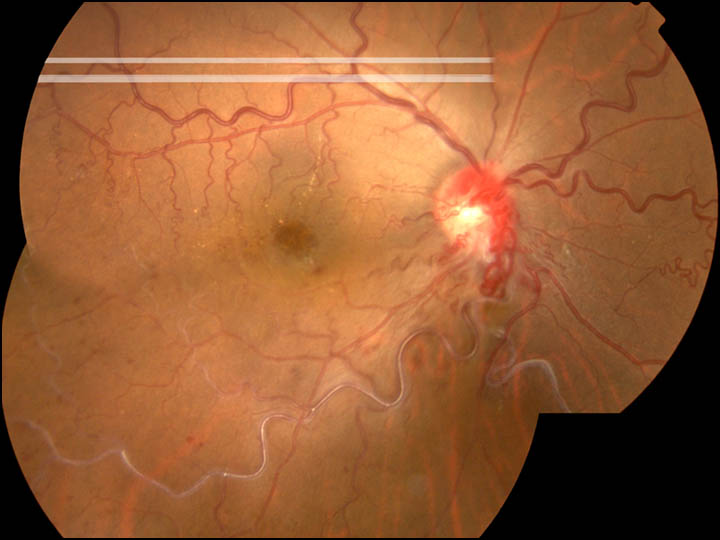 |
This case showes the macular cystic degeneration associated with impending occlusion of inferotemporal branch vein secondary to arteriovenous communications. |
- arterial loop on the optic disc
- venous loop adjacent to the optic disc
- mild malformations
congenital retinal macrovessels(先天網膜血管異常
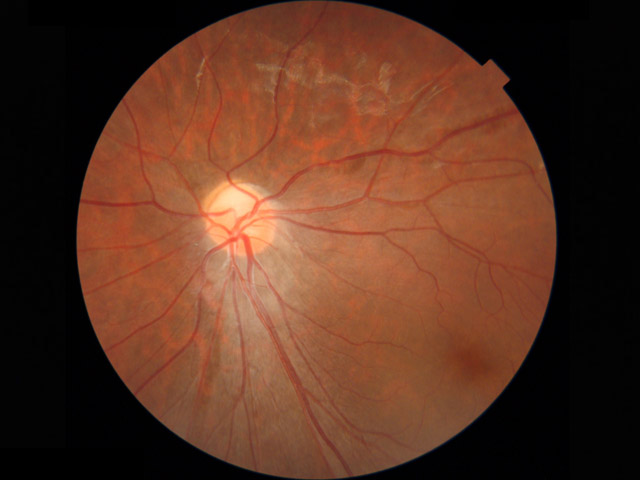 retinal hypovasculopathy(網膜血管形成過程の欠陥に因るカテゴリー
retinal hypovasculopathy(網膜血管形成過程の欠陥に因るカテゴリー
Coats
Norrie disease
familial exudative vitreoretinopathy(FEVR
fascioscapulohumeral muscular dystrophy(FSHD
the osteoporosis pseudoglioma syndrome
周辺部網膜血管の成長不良,既存血管の機能不全や異常拡張,が共通病変となる.
CRB1遺伝子(1q31.3)あたりがあやしいらしい.
(CRB1:網膜色素変性や Leber先天盲関連であるが ・・・・・
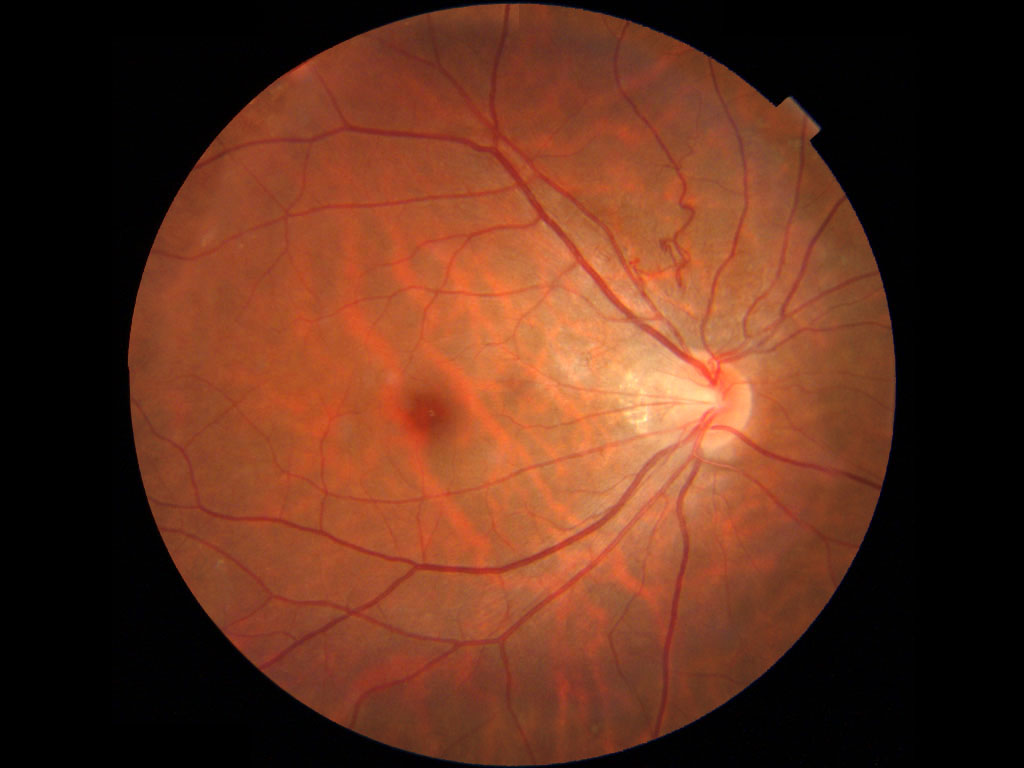
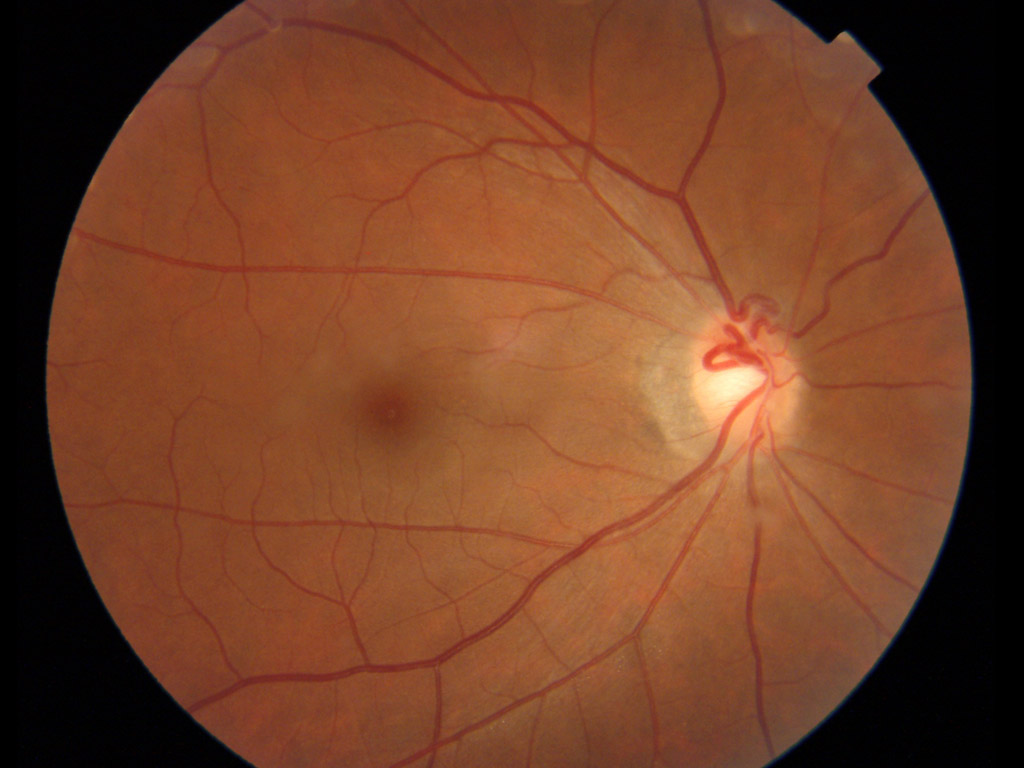
右写真では周辺網膜血管の画像情報はないが,異常回旋のほかmildながら複数の形成異常の合併があり,次のFEVR症例と共通する所見のようにみえる.
☑FEVR
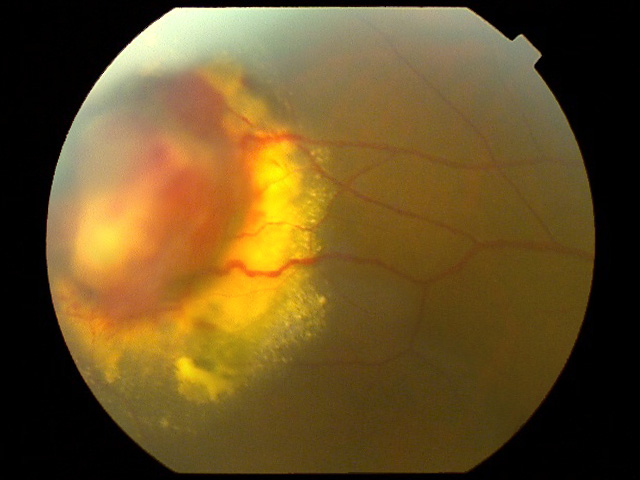
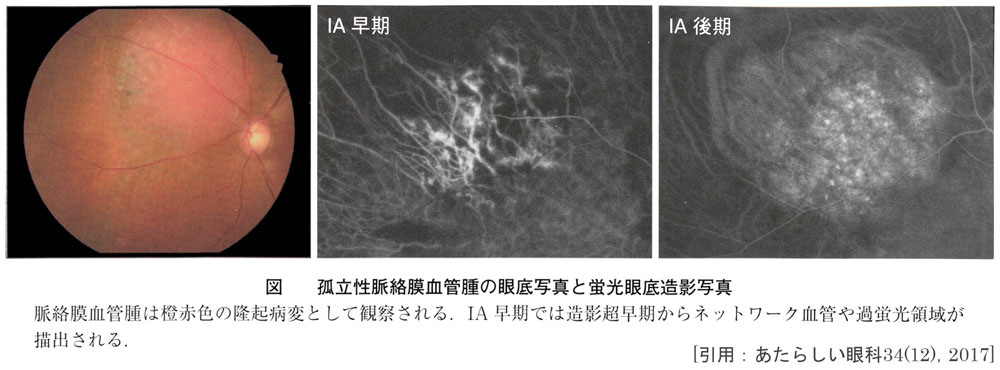
- 網膜血管増殖性腫瘍 retinal vasoproliferative tumors
または 後天性網膜血管腫





{kind=link}
{kind=link}