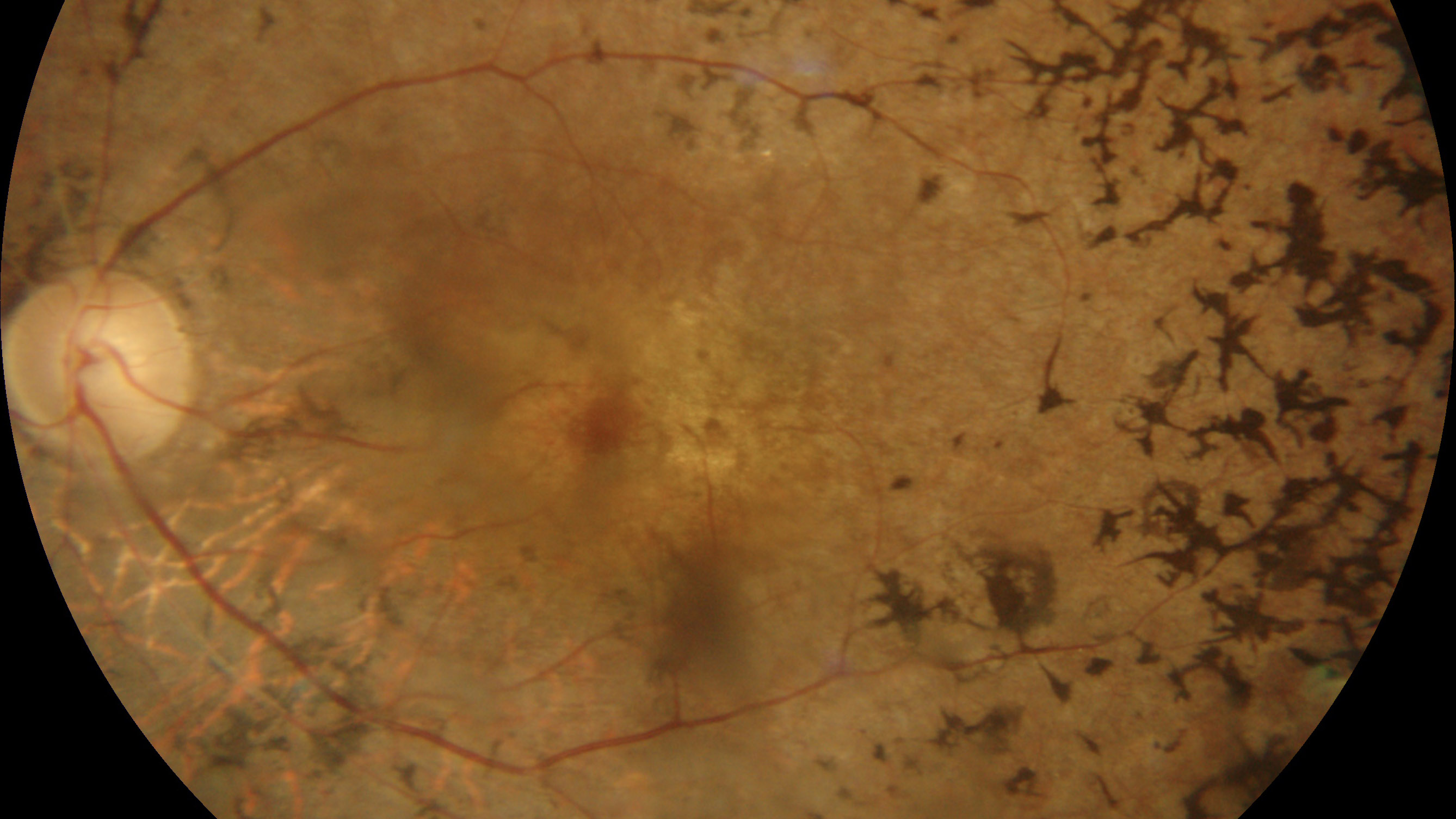
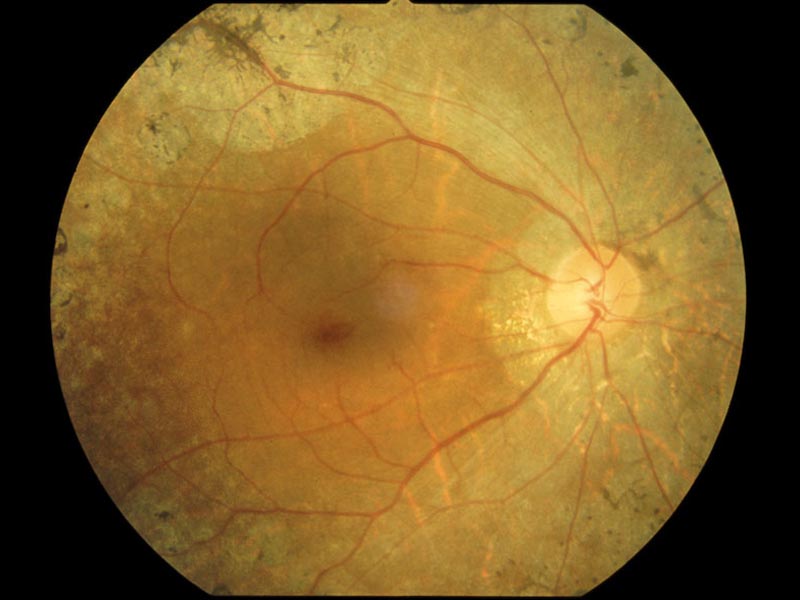
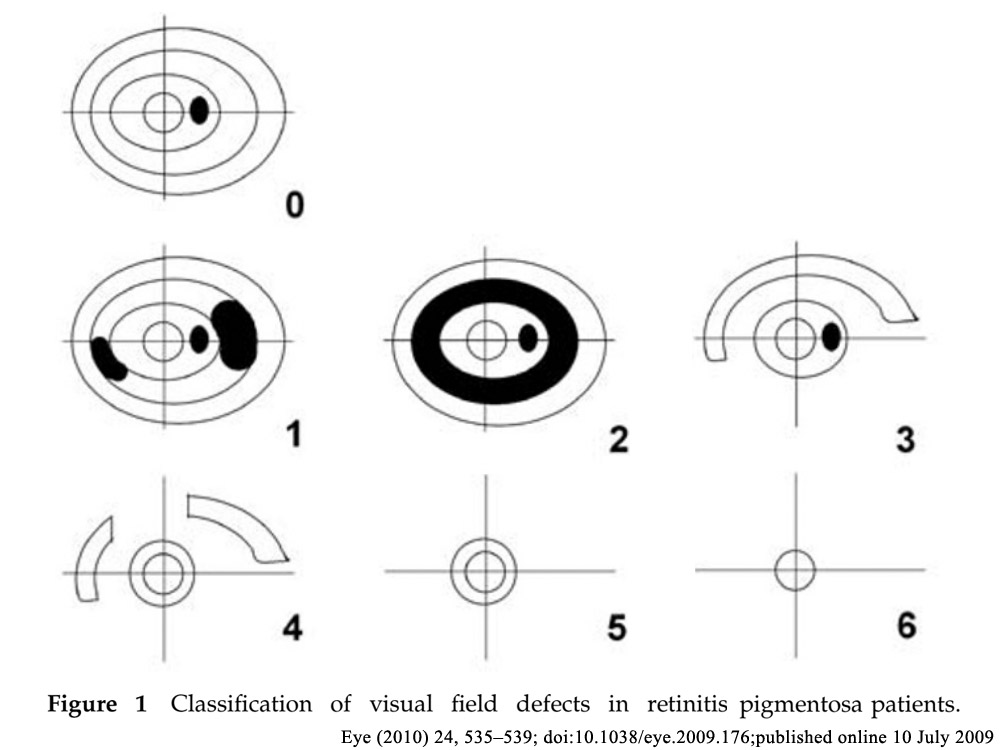
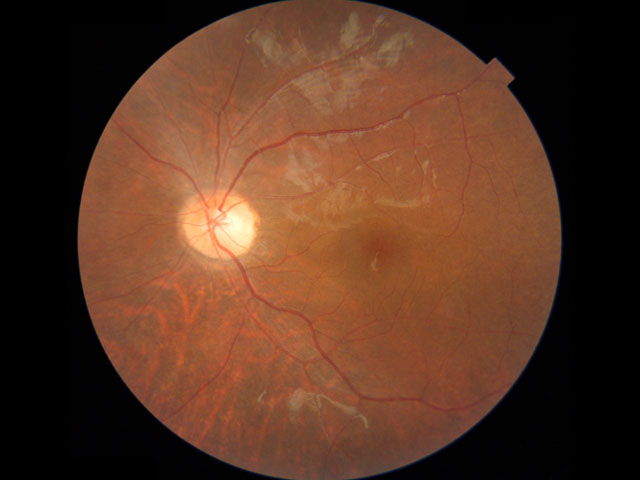
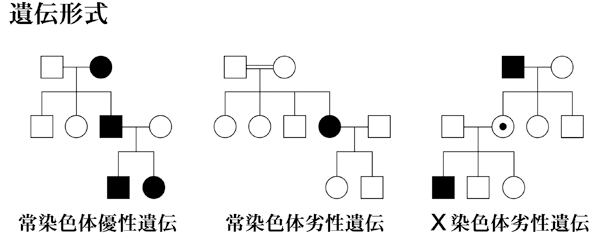
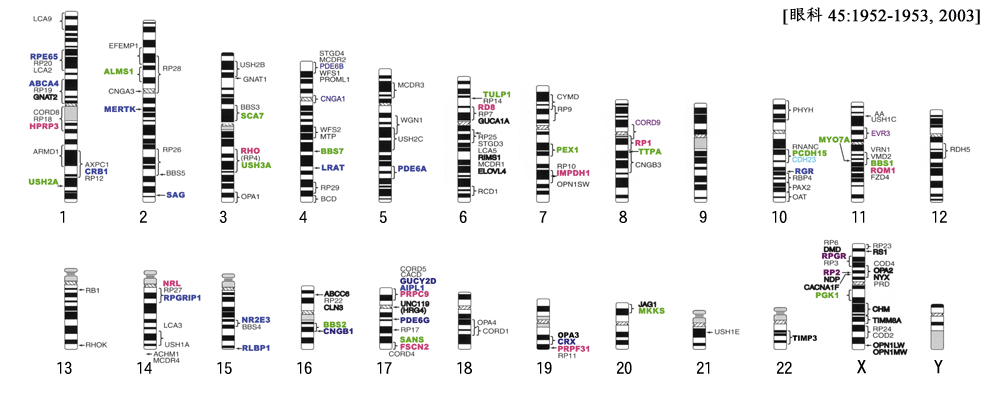
| 1) 短腕: | RPE65(常染色体劣性網膜色素変性,Leber先天盲),ABCA4(黄斑変性,Stargardt病,錐体-杆体ジストロフィ,常染色体劣性網膜色素変性),COL11A1(Stickler症候群) |
| 長腕: | PRPF3(常染色体優性網膜色素変性),CRB1(常染色体劣性網膜色素変性,Leber先天盲),USH2A(Usher症候群?型),ミオシンⅦA(Usher症候群Ⅰ型),ミオシリン(開放隅角緑内障),
ほかに全色盲,Stargart病 ? |
| 2) 短腕: | EFEMP1(黄斑変性,黄斑ドルーゼン),CYP1B1(先天緑内障), |
| 長腕: | CNGA3(全色盲),SAG(アレスチン;日本人小口病,常染色体劣性網膜色素変性),CERKL(常染色体劣性網膜色素変性), |
| 3) 短腕: | GNAT(先天停止性夜盲),ATXN7(SCA7;常染色体優性脊髄小脳変性症), |
| 長腕: | RHO(ロドプシン;常染色体優性/劣性網膜色素変性,停止性夜盲),USH3A(Usher症候群?型),OPA1(常染色体優性視神経萎縮) |
| 4) 短腕: | PDE6B(錐体-杆体ジストロフィ,先天停在性夜盲,常染色体劣性網膜色素変性),CNGA1(常染色体劣性網膜色素変性) |
| 長腕: | LRAT(常染色体劣性網膜色素変性),CYP4V2(クリスタリン網膜症),PITX2(Rieger症候群),ANT1(進行性外眼筋麻痺) |
| 5) 短腕: | |
| 長腕: | GRM6(先天停在性夜盲),MASS1(Usher症候群Ⅱ型),PDE6A(常染色体劣性網膜色素変性),WDR36(開放隅角緑内障) |
| 6) 短腕: | RDS(網膜色素変性,錐体ジストロフィ),GUCA1A(錐体-杆体ジストロフィ),FKHL7(iridogoniodysgenesis,Rieger症候群),TULP1(常染色体劣性網膜色素変性,Leber先天盲),COL11A2(Stickler症候群) |
| 長腕: | RIMS1(錐体-杆体ジストロフィ),ELOVL4(錐体ジストロフィ,黄斑変性),PEX7(Refsum病),
ほかに黄斑ジストロフィ ? |
| 7) 短腕: | RP9(常染色体優性網膜色素変性), |
| 長腕: | PEX1(Refsum病),IMPDH1(常染色体優性網膜色素変性),OPN1SW(常染色体優性第三色覚異常) |
| 8) 短腕: | |
| 長腕: | RP1(常染色体優性網膜色素変性),PXMP3(Refsum病),CNGB3(全色盲),
OPA6(常染色体劣性視神経萎縮) |
| 9) 短腕: | KCNV2( |
| 長腕: | PRPF4( |
| 10) 短腕: | PHYH(Refsum病),OPTN(開放隅角緑内障) |
| 長腕: | PCDH15(Usher症候群Ⅰ型),CDH23(Usher症候群?型),RGR(常染色体劣性網膜色素変性),PAX2(コロボーマ),OAT(脳回状網脈絡膜萎縮),HtrA1(加齢黄斑変性) |
| 11) 短腕: | USH1C(ハルモニン;Usher症候群Ⅰ型),ほかに家族性硝子体網膜症 |
| 長腕: | ROM1(常染色体優性網膜色素変性),VMD2(ベストロフィン;卵黄様黄斑変性),BBS1(Bardet-Biedl症候群),LRP5(FEVR),MYO7A(Usher症候群Ⅰ型),FZD4(FEVR),C1QTNF5(黄斑変性),
ほかに,チロシナーゼ(眼・皮膚白子症) |
| 12) 短腕: | |
| 長腕: | COL2A1(Stickler症候群),RDH5(白点状眼底,錐体-杆体ジストロフィ) |
| 13) 短腕: | |
| 長腕: | RB1(網膜芽細胞腫),GRK1(ロドプシンキナーゼ;欧米人小口病) |
| 14) 短腕: | |
| 長腕: | NRL(常染色体優性/劣性網膜色素変性),RPGRIP1(常染色体劣性Leber先天盲),RDH12(常染色体劣性Leber先天盲),ほかに,杆体一色型色覚 |
| 15) 短腕: | |
| 長腕: | NR2E3(常染色体劣性網膜色素変性,enhanced S-cone症候群,Goldmann-Fabre病),RLBP1(常染色体劣性網膜色素変性),フィブリリン(Marfan症候群) |
| 16) 短腕: | ABCC6(弾力線維性仮性黄色腫,網膜色素線条症) |
| 長腕: | BBS2(Bardet-Biedl 症候群),CNGB1(常染色体劣性網膜色素変性) |
| 17) 短腕: | PRPF8(常染色体優性網膜色素変性),AIPL1(常染色体劣性Leber先天盲),GUCY2D(錐体-杆体ジストロフィ,常染色体劣性Leber先天盲) |
| 長腕: | CA4(常染色体優性網膜色素変性),USH1G(Usher症候群?型),FSCN2(常染色体優性網膜色素変性) |
| 18) 短腕: |
|
| 長腕: | CORD1(錐体-杆体ジストロフィ) |
| 19) 短腕: | |
| 長腕: | CRX(錐体-杆体ジストロフィ,常染色体優性網膜色素変性,常染色体優性/劣性Leber先天盲),PRPF31(常染色体優性網膜色素変性) |
| 20) 短腕: | 網脈絡膜萎縮 |
| 長腕: | |
| 21) 短腕: | |
| 長腕: | Usher症候群Ⅰ型 |
| 22) 短腕: | |
| 長腕: | TIMP3(Sorsby黄斑ジストロフィ) |
| 23) 短腕: | RS1(若年網膜分離症),RPGR(X染色体劣性網膜色素変性),NDP(Norrie病),CACNA1F(不完全型停止性夜盲),RP2(網膜色素変性),OA1(眼白子),NP(FEVR) |
| 長腕: | CHM(コロイデレミア),TIMM8A(遺伝性視神経萎縮),OPN1(色覚異常),
錐体ジストロフィ |
| mitochondrion | LHON(Leber病),
KSS(KearnsⲻSayre症候群) |