- 眼球の先天異常
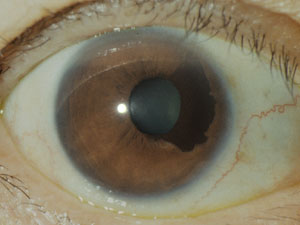
- 無眼球 anophthalmos / anophhalmia
最強度の眼先天異常となる.組織学的に眼球の発生を認めない真性または原発性無眼球は,胎生2週以前で眼小窩の形成に失敗したもので,一次眼胞は発生を中止し(眼胞無形成),脳主要奇形を含む異常発生により,おおむね生存できない.外胚葉性の組織が少量でも残っていれば,「高度小眼球」または「臨床的無眼球」に相当する.
これらでは,眼窩は小さく瞼裂狭小となる.眼瞼,結膜囊,涙器などは存在するものの,結膜囊を含め,眼窩腔・前頭骨・顔面骨の発育遅延となる.片眼では顔面が非対称となるため,整容的対応をする.
・原発無眼球:4週の眼胞不形成を含む.
・続発無眼球:前脳の発生が抑制され,眼球の発生がないもの.
- 単眼症 cyclopia / synopsia
両側の眼の要素が部分的あるいは完全に癒合して顔面正中部に1つの眼が作られた状態(正中眼 median eye)で,両眼の完全な癒合を単眼症 cyclopia,部分的に癒合したものを合眼症 synophthalmia と呼ぶ.通常,前脳胞が左右の大脳に分割しない全前脳胞症に合併してみられ,神経堤の形成障害と関連して発生する.出生前の超音波診断で発見されることもあり,劣性遺伝のほか多くが13trisomy などの染色体異常に関連し,長期生存は望めないとのことである.
(ギリシャ神話では Cyclops;Kýklōps;Κύκλωψ=„round eye“)
- 眼囊胞 congenital cystic eye,眼窩眼瞼囊胞
眼胞の陥凹が失敗,あるいは閉鎖しなかった眼杯裂の端が囊胞状に拡大したもの.すなわち網膜剝離状態である.
胎生裂の閉鎖不全が高度で,増殖した未分化な内板組織が胎生裂部から周囲組織に突出して囊胞を作り,眼球自体の発育も障害されて(コロボーマ性)小眼球となっている.囊胞は眼球の下方に形成され,下眼瞼の腫瘤を形成してくることが多い.囊胞が増大する場合には,眼球摘出に至ることがある.
- 潜在眼球 cryptophthalmos
眼瞼の欠損=瞼裂の欠如ということで,眼球は皮膚で覆われる.眼球は不完全な小眼球にとどまり,角膜・結膜もみられない.睫毛・眉毛も形成異常となる.
潜在眼球症候群となると常染色体劣性遺伝.
- 真性小眼球 nanophthalmos
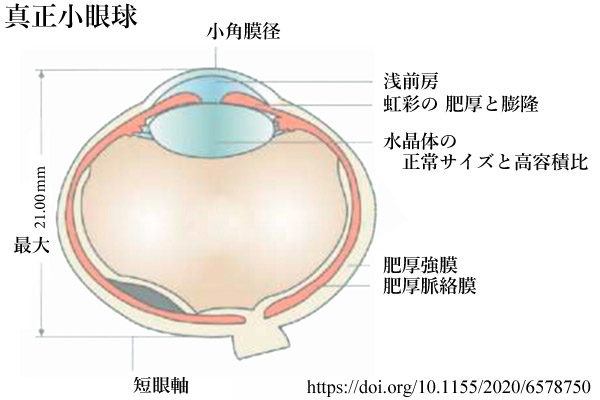
構造は正常でありながら,眼球の大きさが小さいもの.眼軸長は,年齢によるが 14.0〜20.5mmと正常の0.87以下である.眼球体積は正常の2∕3以下である.
臨床的には小眼球症として,眼軸:21mm未満(成人)・19mm未満(1歳),角膜径:10mm以下(成人)・9mm以下(乳児)が目安になる.
結合織成分の異常によって強膜の肥厚があり,発育障害によって眼球の拡大ができない.高度の遠視となるほか多くは視力不良で,黄斑形成不全,網膜血管蛇行,偽乳頭浮腫などを伴うことがある.半数例が両眼性である.
水晶体体積は正常眼と同等である.正常眼での水晶体体積の割合は3〜4%であるが,本症では10〜32%ほどになる.これにより水晶体は相対的に大きく,水晶体因子 として相対的瞳孔ブロック・閉塞隅角緑内障発症の素因となる.肥厚強膜(異常コラーゲン,ムコ多糖の変化)と
脈絡膜の異常な発達により渦静脈の流出障害が起きると,uveal effusion(ぶどう膜滲出)の素因となる.
常染色体優性遺伝 (NNO1,NNO3)あるいは劣性遺伝 (NNO2;MFRP遺伝子<11q23.3> ),SOX2, PAX6, RX, CHX10など.他の身体的異常は伴わない.
nános(νάνος)=dwarf
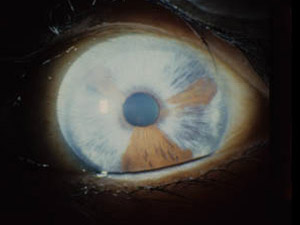
- 小眼球 microphthalmos
臨床的無眼球症,極度小眼球症という重症のものから僅かに眼球が小さいものまで様々な程度がある.構造異常があり眼球発育が障害されたもので,多くは眼杯裂閉鎖不全による.胎生8週付近に原因があれば,(単純)小眼球ということで先天白内障や第一次硝子体過形成遺残など種々の眼組織発生障害を伴う.原因が6週付近にあれば眼杯裂は閉鎖できず,重度のコロボーマ性小眼球となる.4週付近での発達障害では眼球退化物の存在,つまり眼胞の形成までであれば(組織学的には)小眼球であるが,臨床的には無眼球となる.両眼性>片眼性.
染色体異常(13trisomy,cat's eye syndrome,4q-syndrome,etc )が確認されることがあるように,早期の形成障害には遺伝的要因があり,他の身体的異常を伴う.
単純小眼球では感染性病原体(風疹ウイルス,単純ヘルペスウイルス,トキソプラズマ原虫,・・・ )による.
眼球自体の大きさは正常範囲で,角膜径のみの縮小は(狭義)小角膜となる.
- 欠損 coloboma(-詳細は3 で-)
コロボーマの程度や部位に応じて視機能が障害され,脈絡膜欠損では成長の過程で網膜剝離などの合併症を生じてくることがあるので,定期的な経過観察が必要である.
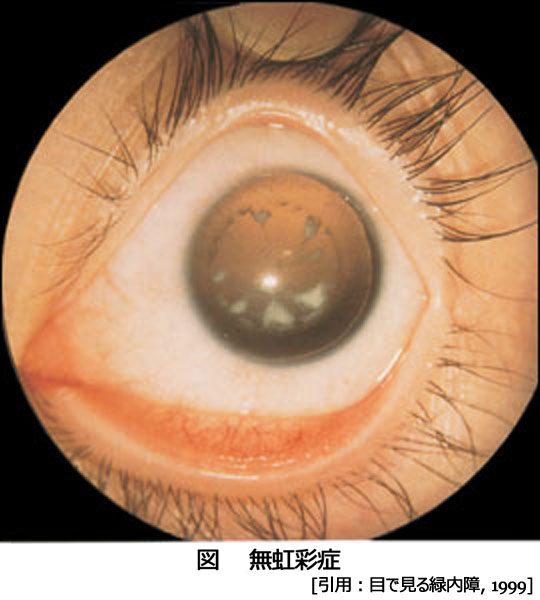
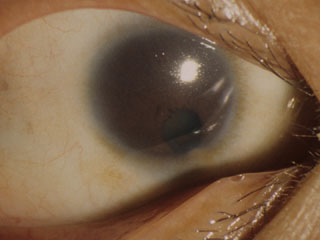
無虹彩(異型虹彩欠損:iridotrabeculodysgenesis)
原 因:PAX 6 の変異あるいは発現の不調により神経外胚葉・眼杯縁の発生停止となったもの.名の通り隅角異常のほか,網膜では黄斑低形成・視神経低形成を伴う.
60〜90%が両眼性.
● I,常染色体優性遺伝:他の全身異常を伴わない型.
●II,常染色体優性遺伝:WAGR症候群.
●III,常染色体劣性遺伝:精神遅滞,小脳性運動失調を伴う型.
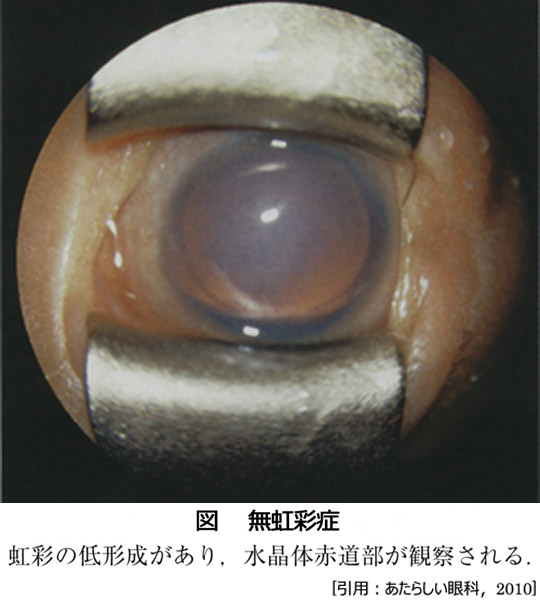
組織構造:毛様体筋の手前に色素上皮の形成不全を伴う小さな虹彩基質根部としての痕跡がある.これにより低形成 iris hypoplasiaに相当し,眼球正面からは虹彩は見えず,毛様体突起が見えることになる.実際の虹彩異常はさまざまで,虹彩表面の部分的萎縮ほどの軽症もある.なお真の虹彩欠損はまれとされる.80%に小眼球を合併する.
症 状:75%に視神経の形成不全を伴い,黄斑発達障害にも関係するとされる(中心小窩無形成は,白皮症のそれと同等とのこと).
これらにより,羞明 photophobia,低視力 decreased vision,弱視 amblyopia,斜視 strabismus,などを示す.
合 併:白内障は50〜85%に発生し,10〜20代で手術を必要とすることが多い.さらに先天緑内障を50〜75%で発症する.水晶体偏位 ectopia lentisは50%,ほかに小水晶体 microphakiaがみられることがある.胎生環 arcus juvenilisによる角膜混濁のほか,本症は角膜上皮細胞が過剰に入れ替わることから,
palisades of Vogt が消失する.成長につれ輪部機能不全/角膜上皮幹細胞疲弊症をきたし,パンヌスを形成し,混濁が進行する.Meibom腺異常も合併する.
PAX6 は神経系の発生にも関与することで,先天眼振congenital nystagnusは原発症状と考えられている.
遺 伝:遺伝性は2∕3,孤発性が1∕3といわれる.
家族性 familial:遺伝性PAX6変異により優性 dominamt 形式をとる.
孤発性 sporadic:妊娠の比較的早期に変異をきたすと考えられる(両親は正常).
WAGR 症候群(Wilms' tumor,Aniridia,Genitourinary anomalies,mental Retardation ):PAX6近傍の遺伝子を含む変異による(Chromosome 11p13 deletion syndrome).腎 Wilms腫瘍の原因遺伝子 WT 1は位置が近いため併発.希で孤発性らしい.
Gillespie症候群:PAX6変異ではないと考えられている(IP₃イノシトール三リン酸受容体遺伝子,常染色体劣性).大きな虹彩根部痕跡(scalloped と形容)に全身症状(精神発達遅延,小脳性失調)を伴う.希.
✓⌘ aniridiaは先天性や外傷性で一般的に用いる.
irideremiaとすると先天性を意味するそうである(erēmia;ερημια = absence,desert, wilderness).
✓⌘
後部胎生環:Schwalbe線の隆起.こちらで.
前部胎生環≒若年環 ⇔ 老人環 とは別物.
✓⌘ ここで云う『欠損』と次のコロボーマの『欠損』とを,混同しないこと.
虹彩が無い → 欠損とは云えないので “異型” を付ける.
-
小瞳孔 microcoria
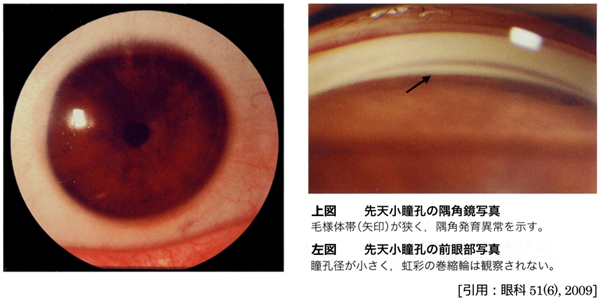
不明瞭な虹彩紋理で,径2mm以下となる.対光反応や近見反応がない.瞳孔散大筋の分化異常により,通常は両眼性.隅角発育異常を伴い,高率に緑内障を発症する(遅発型発達緑内障).
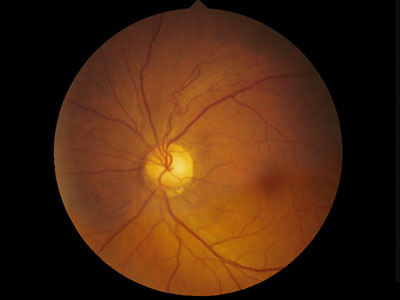
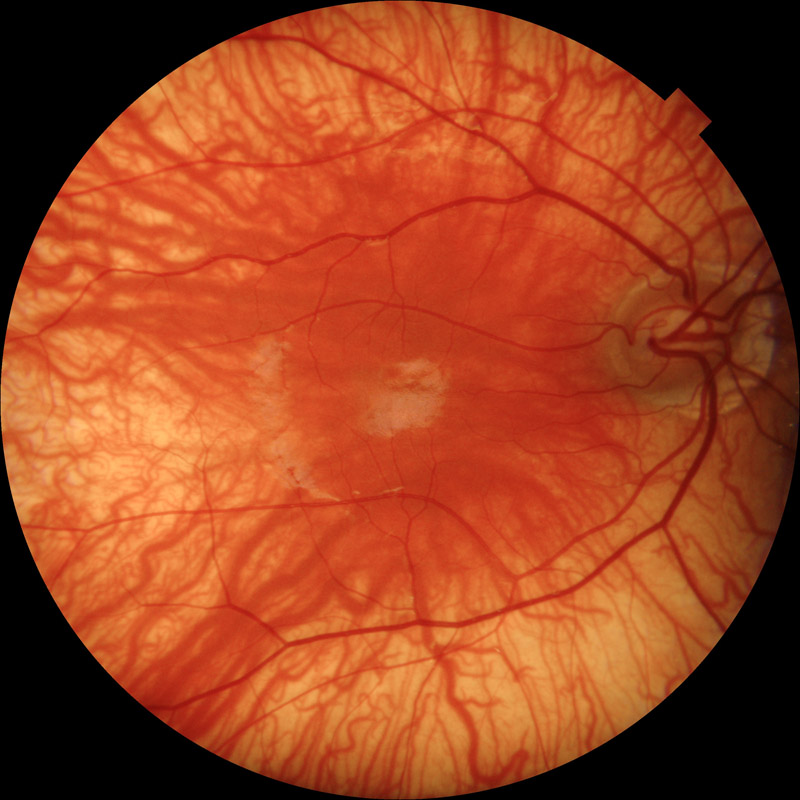
ぶどう膜欠損:uveal colobomas
【 眼底写真 】
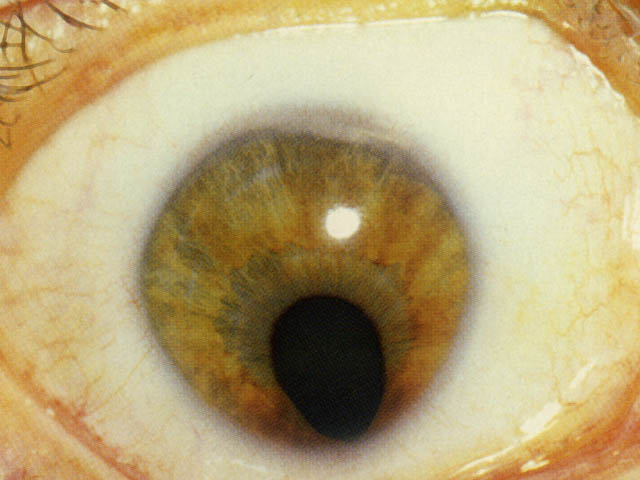
原因:(狭義)コロボーマは眼杯裂の融合・閉鎖の障害による.外胚葉の責任でぶどう膜組織の誘導が失敗したもので,ぶどう膜欠損である.欠損部では網膜色素上皮細胞層と脈絡毛細血管板が発達しない.強膜も発育が悪く,網膜も形成不全となる.
組織構造:構成組織の一部が欠損する状態がコロボーマである.ここでは特に “ぶどう膜組織の一部欠損”を指している.定型的には鼻側下方に現われ,虹彩から視神経までの完全な欠損から,限局した小さな欠損まで各種の移行型がある.
形態:眼杯裂前端の虹彩,あるいは最後部の乳頭隣接網膜で高頻度に現われる.中央から融合あるいは閉鎖が始まり,前方向と後方向へ進行するためである.
発生部位により虹彩欠損,毛様体や毛様小帯の欠損,脈絡膜欠損,視神経乳頭欠損などに分ける.
- 瞳孔偏位 corectopia:最も軽度な眼杯裂(遠位端)の閉鎖障害.
- 虹彩欠損(虹彩コロボーマ)あるいは鍵穴(孔)瞳孔:虹彩単独での形態.虹彩根部は萎縮状態の痕跡がある場合が多い(partial coloboma).完全欠損では毛様体部までを含む(total coloboma).
- 毛様体欠損(毛様体コロボーマ):Zinn小帯の欠損を伴うと 水晶体欠損(lens notching)をきたす(ここでも,水晶体欠損=無水晶体,ではない).
- (網)脈絡膜欠損((網)脈絡膜コロボーマ):基本的に,網膜色素上皮・Bruch膜・脈絡毛細血管板が欠落し,透見されるのは強膜.異常網膜が存在し,よく穴が開く.
- 黄斑欠損:黄斑組織の欠損
- 視神経乳頭コロボーマ:乳頭組織の欠損,異常血管.
- 視神経乳頭下方コーヌス:最も軽度な眼杯裂(近位端)の閉鎖障害.
- 下方ぶどう腫:軽度の網脈絡膜萎縮よって部分的な豹紋状眼底と,その境界がやや変性傾向となる.傾斜乳頭症候群での網脈絡萎縮部がそれ.OCT上の菲薄脈絡膜のひとつに挙げられている.
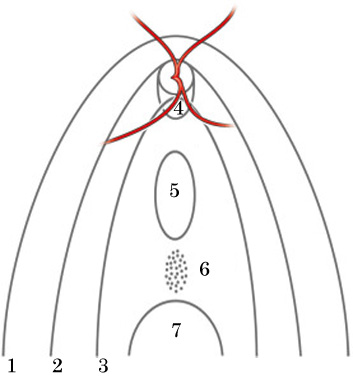
► Ida Mann(1937)は,タイプ分けを提唱している.
➀乳頭を超えた範囲,➁乳頭を超えない上端まで,➂乳頭下端には届いていない,➃乳頭部のみ(下方コーヌスを含む),➄孤立性のコロボーマ,➅未発達の色素沈着,➆周辺に存在.
である.
► 多くは小眼球(colobomatous microphthalmos)である.視神経乳頭小窩 optic disc pit,朝顔症候群 morning glory syndrome,傾斜乳頭症候群 tilted disc syndrome,先天囊胞眼 congenital cystic eye などが同じスペクトラムにある.
► コロボーマ(ぶどう膜欠損)の程度や部位に応じて視機能が障害され,脈絡膜欠損では成長の過程で網膜剝離や網膜下新生血管などの合併症を生じてくることがあるので,定期的経過観察が勧められる.
► 遺伝:常染色体優性の型
その他は,22番染色体,18番染色体,13番染色体,8番染色体,4番染色体,などの異常による症候群に合併.
PAX2異常による腎コロボーマ症候群(腎奇形と特に朝顔症候群),
【  PAX 】
PAX 】
など
► CHARGE 症候群:
・C:coloboma of the eye 眼のコロボーマ(眼の構造の部分欠損),
・H:heart defect 先天性心疾患,
・A:atresia of the choanae 後鼻孔閉鎖,
・R:retardation of growth and/or development 成長や発達の遅れ,
・G:genital and urinary abnormalities 外性器や尿路系の異常,
・E:ear malformation and/or hearing loss 耳の形態異常や聴力障害,
8番染色体上のCHD7遺伝子の機能不足による多発奇形,常染色体優性疾患
► COACH 症候群:
・C:小脳虫部低形成 cerebellarvermis hypoplasia,
・O;精神遅滞 oligophrenia,
・A:小脳失調 ataxia,
・C:脈絡膜欠損 coloboma,
・H:肝線維症 hepatic fibrosis
常染色体劣性遺伝疾患で,一次繊毛という細胞小器官の形成に関与している.
【
繊 毛 】
► 歯科・口腔外科領域の “口唇口蓋裂” に対応する病態である.
-
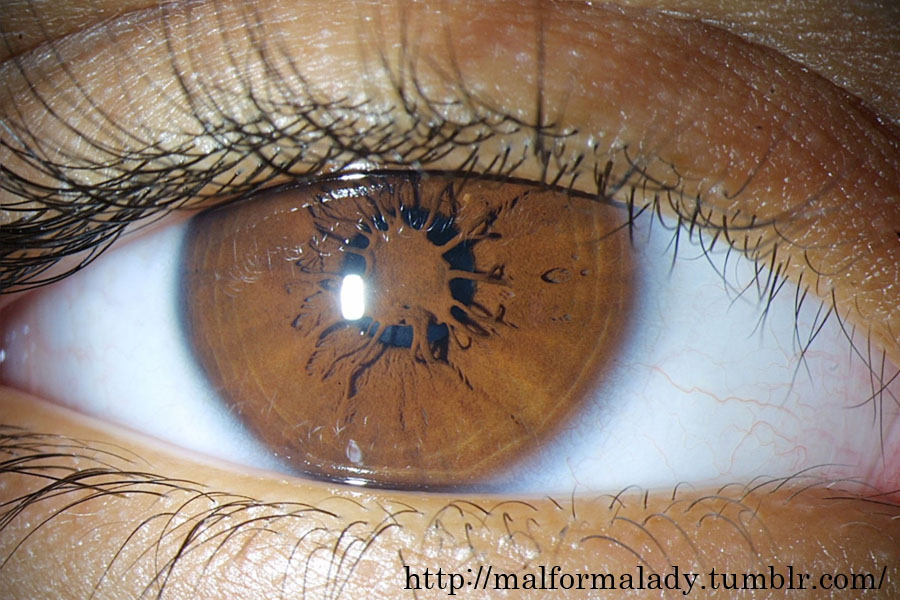
瞳孔膜遺残 persistent pupillary membrane
水晶体胞の前面は,虹彩原基から伸びた血管 tunica vasculosa lentisで栄養される.瞳孔膜である.これは硝子体動脈の延長にある.瞳孔膜血管の基は,虹彩の捲縮輪として残っている.言い換えると,捲縮輪の中にある小虹彩動脈輪は瞳孔膜血管から出来る.瞳孔膜遺残は捲縮輪から連続していることになる.
瞳孔膜は,妊娠6か月目にマクロファージの貪食作用によって退縮し,妊娠8か月目までに完全に消失する.出生後に残った瞳孔膜も,生後1年でかなり萎縮する.瞳孔膜の吸収に失敗すると,瞳孔膜遺残につながる.
角膜後面への癒着状態(Duke᠆Elder分類によると type4)のひとつ(虹彩形成不全)が Rieger's anomaly となる.
【 ☟
前眼部形成異常 】
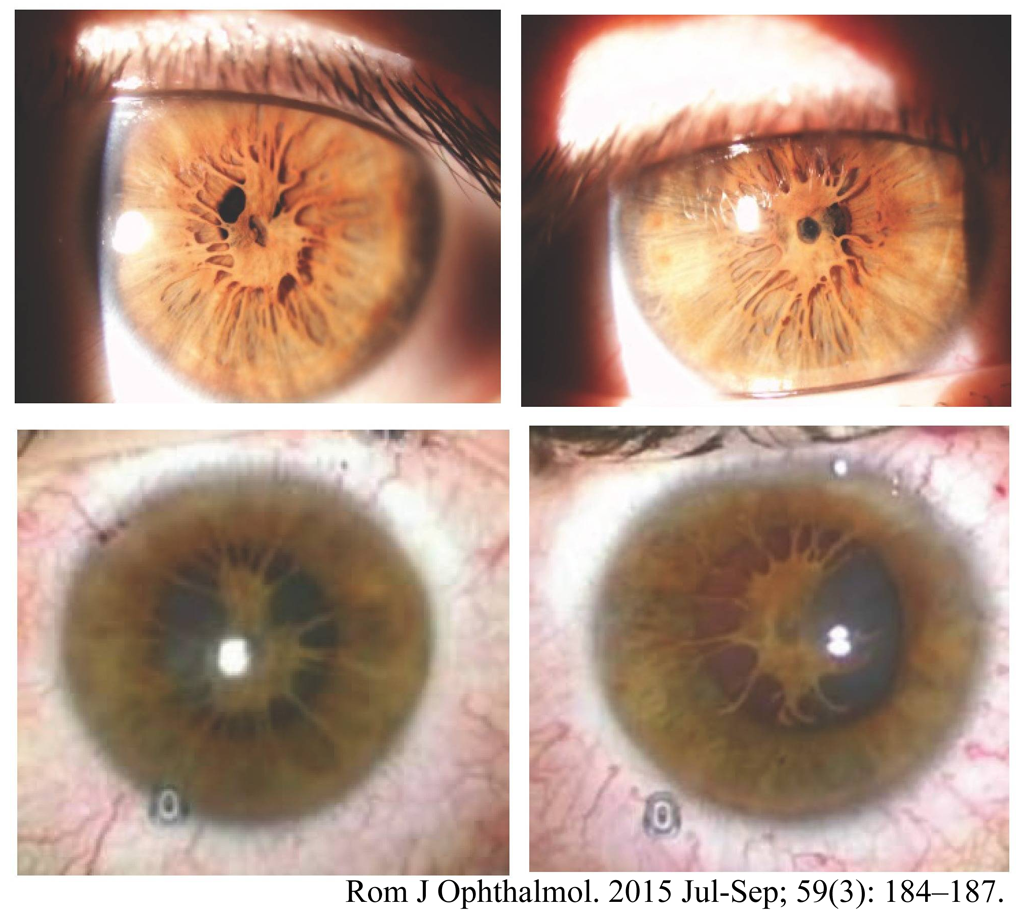
-
accessory iris membrane または iris duplication
間葉組織の過形成で,筋組織のない擬似瞳孔がある.臨床的には,瞳孔膜遺残との区別は相当にむつかしそうである.
-
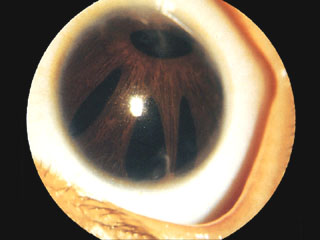
多瞳孔 polycoria
右 写真
-
瞳孔偏位 corectopia
先天性ではコロボーマの一形のほか,Axenfeld᠆Rieger異常,等
後天性には,ICE症候群(原因は先天性であるが経年変化で進行する.「part Ⅱ」で),
時に合併症として・・・・
kóre(κόρη)=pupil,ektopos(εκτόπος )=displaced
- 前眼部間葉異発生
先天角膜混濁(先天性白斑)などの角膜形成異常および前房領域の異常などを含み,神経堤細胞(二次性間葉細胞)の,遊走・増殖・分化の障害と考えられている.
【
参考 】
- 遊走の障害:先天緑内障,後部胎生環emblyotoxon posterior,Axenfeld,Reiger,Peters,etc
【
参考 】
- 増殖の障害:特発性虹彩萎縮,Chandler,
ICE症候群
- 分化の障害:いくつかの角膜内皮ジストロフィ
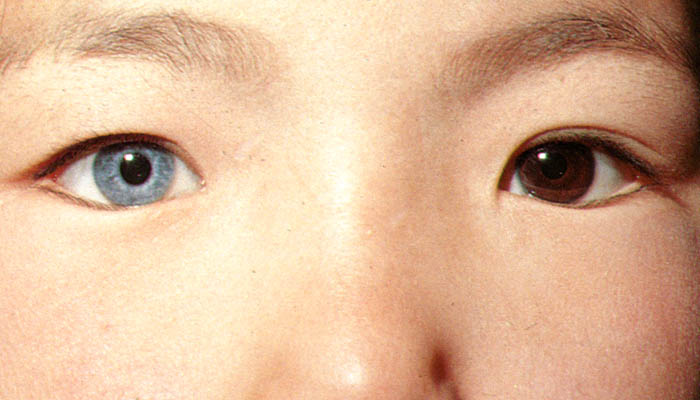
- 虹彩異色 iris heterochromia
虹彩色に左右差がある状態と,一眼の虹彩に部分的色ムラが生じる状態とがある.
- heterochromia iridis(binocular heterochromia)
❶ hyperchromia:濃色.色素の含有量の増加あるいは色素細胞数の増加.
・先天性:多量の色素を持つメラノサイトの増加.いわゆるメラノーシス.
例;太田母斑.線維柱帯内の色素は緑内障発症の因子となる.
・後天性:鉄さび症,色素沈着,細胞浸潤,血管新生,でみられる.緑内障治療薬(ラタノプロスト)の副作用でも発生.
❷ hypochromia:淡色.メラノサイトの減少と色素の不足.
・先天性:
○単純型.単なる低形成によるもの.
○Horner 症候群
交感神経の脱神経・作用不全により,同側の色素が減少するもの.縮瞳・眼瞼下垂(Müller筋の緊張不足)・眼球陥凹 が 三主徴.また,発汗減少,など.
○Waardenburg 症候群
常染色体優性をとる神経堤細胞異常.虹彩低色素のほか,眼周囲の皮膚・毛髪の低色素に難聴・顔面の形成異常を伴う.
・type1;内眼角開離・涙小点の形成異常がある.
・type2;上を伴わないもの.難聴の頻度が高い.
・そのた
○Fuchs 虹彩異色毛様体炎 ☞
„そのたのぶどう膜炎“ で
・後天性:ぶどう膜炎などで実質の萎縮をきたして
- heterochromia iridum(iris bicolor)
一眼に,色素ムラが生じている.
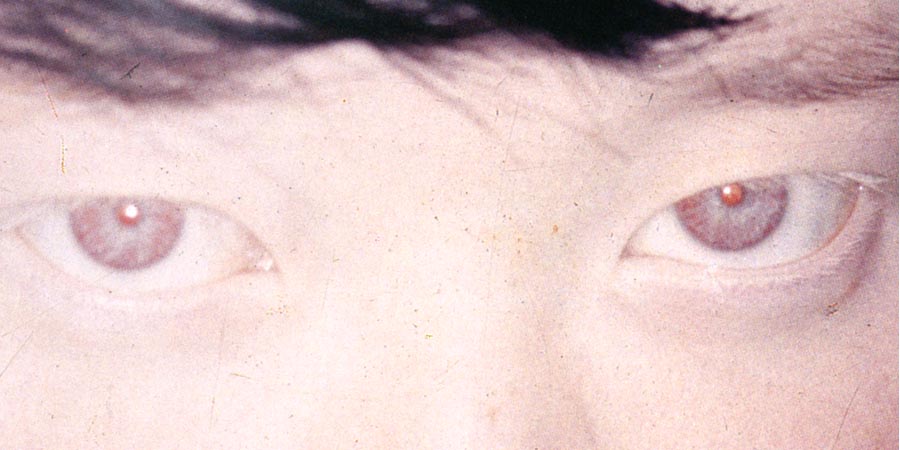
- 白皮症 albinism 白子・アルビノ
メラニンの合成障害(チロシン → メラニンに作用するチロシナーゼの欠乏あるいは利用障害)による.
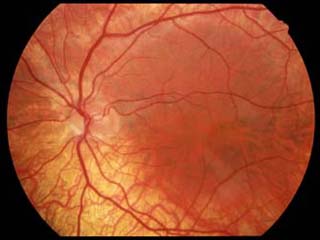
網膜・毛様体・虹彩の色素細胞中には色素の原料顆粒の存在を認めるがメラニンが生成されない.
網膜色素上皮細胞を含む黄斑部中心小窩の形成障害
(黄斑低形成)
があり,視力不良,振子様眼振,羞明,などを示すほか,遠視,斜視(立体視の不良),などを合併する.眼のほか皮膚に対しても遮光手段を要する.眼白皮症では,皮膚や毛髪の色素はおおむね正常.
 ・全身の皮膚・毛髪と眼(眼皮膚白皮症oculocutaneous albinism;OCA):常染色体劣性遺伝
・全身の皮膚・毛髪と眼(眼皮膚白皮症oculocutaneous albinism;OCA):常染色体劣性遺伝
・眼のみ(眼白皮症ocular albinism;OA):X染色体劣性遺伝.臨床での診断上,「男児患者と保因者の母親」,あるいは「女児保因者と発現者としての父親」の確認は診断の助けになる.
Nettleship᠆ Falls型保因者眼底はモザイク様眼底を示す.
Forsius᠆ Eriksson型保因者眼底は正常眼底.
・皮膚のみ(cutaneous albinism):
- OCA type 1;チロシナーゼ遺伝子関連型:TYRチロシナーゼ遺伝子(11q14-21)の変異.日本人で最も多い.約34%
黄斑低形成,視神経軸索の交叉異常がある.これにより低視力(0.1以下),眼振,羞明,斜視を示す.虹彩は明るいblue であるが,時にviolet ないしpink を示すとのこと.
変異の起きかたで‘1a’と‘1b(yellow mutant)’が生じる.
・OCA1a:チロシナーゼ陰性型.チロシナーゼが無いか活性が無く,メラニン合成ができない.日焼けができない.視力は0.1以下.幼小児期の蒙古斑を欠く.
・OCA1b:チロシナーゼ(わずかに,数%の)陽性.生下時には‘1a’位に色素がないが年齢と共に眼球・毛髪・皮膚の色素が増加する.これにより,ときに‘OA’と誤診されたり,経過中に視力が向上することもあるらしい(0.1 → 0.2).確認されている変異部は多数あり,個々により色素量や日焼けの程度が違う.
・OCA1TS:温度感受性(TS:temperature sensitive)チロシナーゼにより,手足の体毛は年齢と共に色素が増加する(上腕・下腿では皮膚温が低く酵素活性が維持され,体幹部は皮膚温が高く酵素が失効する).眼球・皮膚の色素は増えない.
・OCA1MP:最小色素型(MP:minimal pigment)
- OCA type 2;P遺伝子関連型:外国の記述では最も一般的なタイプとなっている.全世界的には約50%,日本人では約8%
網膜・虹彩は正常色素を欠く.黄斑低形成,視神経軸索の交叉異常がある.虹彩は blue ないし gray で光線は透過する.OCA1よりも色素が多め(生後,増加する)で視力もよい(0.2 → 0.5くらいに向上するらしい )が,OCA1bより日焼けが出来ないとのこと.
・15q11-q13変異型:チロシナーゼは正常.メラノソーム膜タンパクの異常でチロシンがメラノソーム内に取り込まれずメラニンが作れない.個々によりメラニン合成能の幅が大きい.
・P蛋白:melanocyte᠆ spacific transporter protein あるいは pink᠆ eyed dilution prorein homolog.皮膚色のメラニン合成に関与し,眼では虹彩色の 茶 ⇔ 青 決定の主役.pinkeye の P かな.
- OCA type 3;チロシナーゼ関連タンパク1遺伝子型:
虹彩は blue か gray,中央が茶系で周辺で薄くなるなど部位により色調が違う,とのこと.
・9p23・Tyrp1(または,TRP-1)遺伝子の変異.チロシナーゼ関連タンパクの異常のため,メラニンが作れないか黒メラニンでなく茶メラニンになる.黄斑低形成・視神経低形成がなく,‘真’の albinism ではないとする研究者もいる.
minimal pigmented type albinism・rufous oculocutaneous albinism を含む.
- OCA type 4;SLC45A2遺伝子型:albinism の臨床症状の特徴を備え,わが国で多い.約27%
・5p13.3・MATP遺伝子(旧称)の変異.膜関連輸送タンパク質遺伝子の異常でOCA2と同様の結果となる.色素合成には幅がある.
- OCA type 5:4q24
OCA type 6:SLC24A5遺伝子(15q21.1),あるいは retinal hypopigmentation
OCA type 7:C10orf11遺伝子
- ocular albinism type 1 :(OA1)眼白皮症の通常の型(Nettleship᠆Falls).
眼底の低色素,黄斑反射の欠如,虹彩の低色素(光透過)が特徴.これらにより斜視,眼振,羞明,立体視の欠如そのたの視力障害,屈折異常を示す.後部胎生環(前眼部の低形成を示唆),角膜径の拡大などを合併.正常ERG
Xp22.3にあるGPR143遺伝子の変異(mutation).欠陥メラノソームのため完成品が出来ないらしい.
- ocular albinism type 2 :(OA2)夜盲・色覚異常(protan)を合併.不完全先天停在夜盲類似のERG
CACNA1F遺伝子の変異.Forsius᠆Eriksson型,あるいはÅland᠆Island型.
- ocular albinism type 3 :(OA3)常染色体劣性遺伝型(AROA).チロシナーゼ遺伝子の変異(14%)か P遺伝子の変異(36%)か,何れの変異でもない例が50%とか.
- ocular albinism with sensorineural deafness :(OASD)
OA1に聴覚障害を合併したもの.
- cutaneous albinism:
頭部では前髪~鼻根部,あごの低色素.加齢により色素が増加.
- melanin:
眼の色素細胞には,神経外胚葉由来の細胞と神経堤細胞由来の細胞がある.前者は眼杯であるから網膜・虹彩毛様体の上皮細胞であり,後者はぶどう膜実質のメラノサイトである.なお後者のメラノサイトは,内耳や皮膚にも分布する.聴力障害を合併しやすいことのようだ.
眼球形成時のメラニン欠乏では,中心窩の形成,視神経(神経節細胞の軸索)の外側膝状体への接続,などがうまく行かないのだそうだ.
- albino:
1.中心窩の低形成をきたす.視交叉部での非交叉線維が少なくなり,モノビジョン優勢で立体視が劣る.
2.虹彩がスケスケで遮光が悪い.translucency,あるいは transillumination などと表現.
3.ERG は‘supernormal’を示す.
4.網膜の光傷害が考えられる.遮光が悪いことに加えてフリーラジカルが増加する.メラニンはフリーラジカルの消去役をしているらしい.
5.角膜上皮細胞と Bowman膜との接着が弱い.フリーラジカルが増加する所為らしい.LASIKで問題になる.
- そのほか:
1.基本的な成長・寿命・知能は,正常範囲.
2.皮膚がんのリスクが増加する.本邦の皮膚科対策は十分周知されているとのこと.ただし悪性黒色腫には注意が必要とされる.
3.他科疾患で albinism を合併するもの(症候型 syndromic.
OCA2とのクロスリンク:PWS(Prader᠆Willi syndrome),AS(Angelman syndrome)
【 ☞
15q11-q13変異 】
OA1とのクロスリンク:X連鎖 ichthyosis,Kallmann syndrome
(X連鎖型,・・・・・
クロスリンクが原因でない場合:HPS(Hermansky᠆Pudlak syndrome―遺伝子座10q23.1-23.3),
CHS(Chédiak᠆Higashi syndrome―遺伝子座1q42.1-42.2),
GS(Griscelli syndrome
4.ロービジョンケア‥
5.社会的ramification…
 日本眼科学会
日本眼科学会
-
そのた
・Brushfield spots:Down syndrome(21トリソミー)での虹彩模様.結合織が多めに存在するもの.アジア系より白人系のほうに高率に出現するとのこと.
・虹彩実質低形成:
【
参考 】
■ PAX6 遺伝子:
PAX6は,神経外胚葉,神経堤細胞,小脳の発生を含む中枢神経の発達などに関与する.眼では,眼杯,神経網膜,水晶体,角膜上皮で遺伝子が発現する.これによりPAX6異常では,
角膜・前房隅角・毛様体・水晶体・黄斑部などに原発病変をきたす.これらが
先天無虹彩,Peters'anomaly,角膜混濁,先天白内障,黄斑低形成,などである.なお,虹彩異常では軽症型あるいは異型ともいえる僅かな萎縮のみなど,幅広い表現型を示すとのことである.また先天眼振についても視力不良や黄斑低形成による続発症状ではなく,原発性に発生したものとされる.研究者は,眼球運動の制御機構が円滑に形成されない,と説明する.
先天無虹彩では 片アリル の機能喪失によって機能遺伝子量が半減
(ハプロ不全)・不足することで発症.
(PAX系の)両アリルが異常であると胎生致死になる,とのことである.
【
PAX 】
■cat eye syndrome:
22番染色体の部分トリソミー.瞳孔の形状による病名.全身的には ・・・・・
■18番染色体短腕あるいは長腕の消失:…
■13番トリソミー:
小眼球や網膜の異形成を合併
【  トリソミー 】
トリソミー 】
■
虹彩色素
:
虹彩の色調は,①色素上皮のメラニン色素,白皮症を除く,②実質内色素細胞の色素顆粒,③光線の吸収量,に依る.
虹彩色の如何に関係なく,メラノサイト数に大きな差はないらしい.メラニン顆粒の数と大きさで,濃淡が決まる.色素が薄く虹彩実質内が透ける状態では,長波長が吸収され短波長が反射することで青い目にみえる.
■過誤腫 hamartoma:
胎児期の発生異常で出来た,結節状あるいは腫瘍状のかたまりの組織で,組織奇形・組織迷入・遺残組織など.本来の構成成分の比率が変わっている状態.母斑や血管腫が代表.
【 過誤腫 】
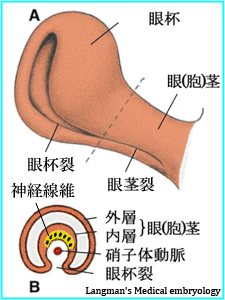
■眼杯裂・瞳孔膜:
疾患病理の解釈には,眼発生の理解が助けになります.
かくして,関連する解剖を見直しをしておくこと.
【  発生 】
発生 】