眼底所見に基づく疾患概念で外因がなく,黄斑機能が両眼性・進行性に低下する病態を総称する.初発部位や主要病変部位による分類で評価するのが一般的である.臨床レベルでは黄斑変性の所見であるが,分子レベルでは網膜全体に及ぶ.
進行型錐体ジストロフィ および 錐体-杆体ジストロフィ cone(ⲻrod)dystrophy;progressive forms
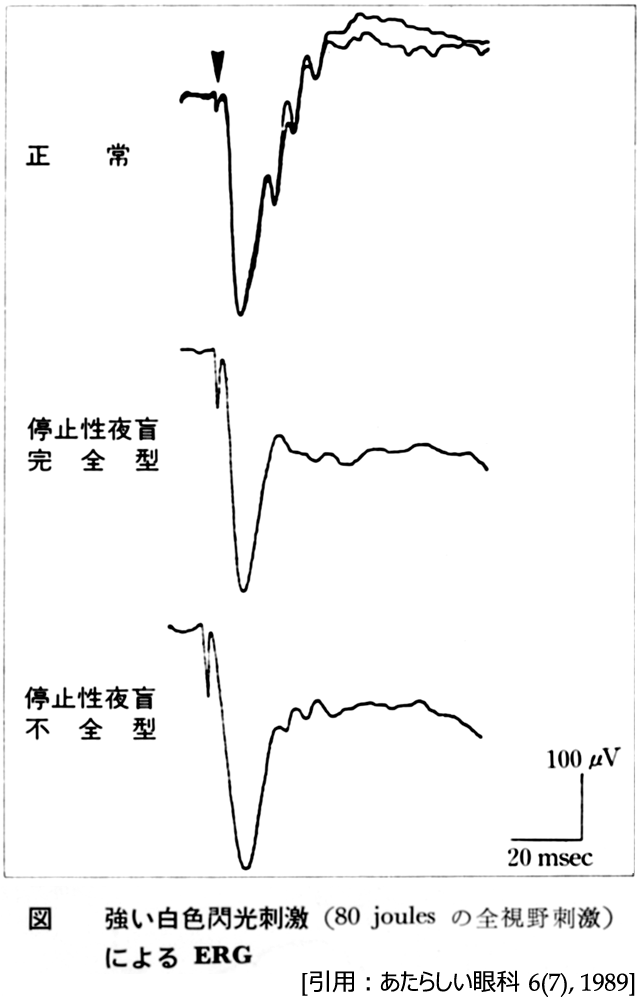
錐体ジストロフィは,錐体機能が杆体機能に先んじて障害される病態である.錐体ERG(30Hzⲻflicker,赤色閃光刺激,明順応下色光ERG,など)の高度異常,黄斑局所ERGの減弱~消失などの機能障害から診断される.端的には全視野刺激ERGにおいて,正常範囲であれば “黄斑ジストロフィ”,明所視の反応低下は “錐体ジストロフィ”,明所視・暗所視の反応低下は “錐体‐杆体ジストロフィ”となる.準正常の暗順応(二次曲線が認められる)を示す.OCTではellipsoid zoneが不明瞭あるいは消失する.
錐体障害が先行することで,10代までに後天性色覚障害を伴う進行性の視力低下を自覚する.羞明も特徴的な症状とされ,経過により夜盲や周辺視野欠損などを引き起こす.要するに定型網膜色素変性とは逆のパターンであるが,杆体反応よりも錐体反応のほうがより高度に障害される状態である.これにより錐体ERGの減衰~消失に加え一次暗順応閾値の上昇をきたす.また昼盲も前面・先行する.なお初期に於いては,検眼鏡・電気生理など正常範囲と捉えられる.
bull's eyeを示す網膜色素変性のバリエーションとして解釈するが,網膜色素上皮変性はわずかであり,眼底が正常色調を示す症例(POC1B関連)も指摘されている.眼底所見の多様性から本症を症候群と解説する研究者が多い.
常染色体劣性(約3/4),優性(約1/5),まれにX連鎖性の遺伝様式がある.
責任遺伝子;GUCY2D,CRX,GUCA1A,など(以上,優性),EYS,PROM1,PRPH2,RPGR1P,KCNV2,ABCA4,ADAM9,など(以上,劣性),RPGR(X連鎖性)など
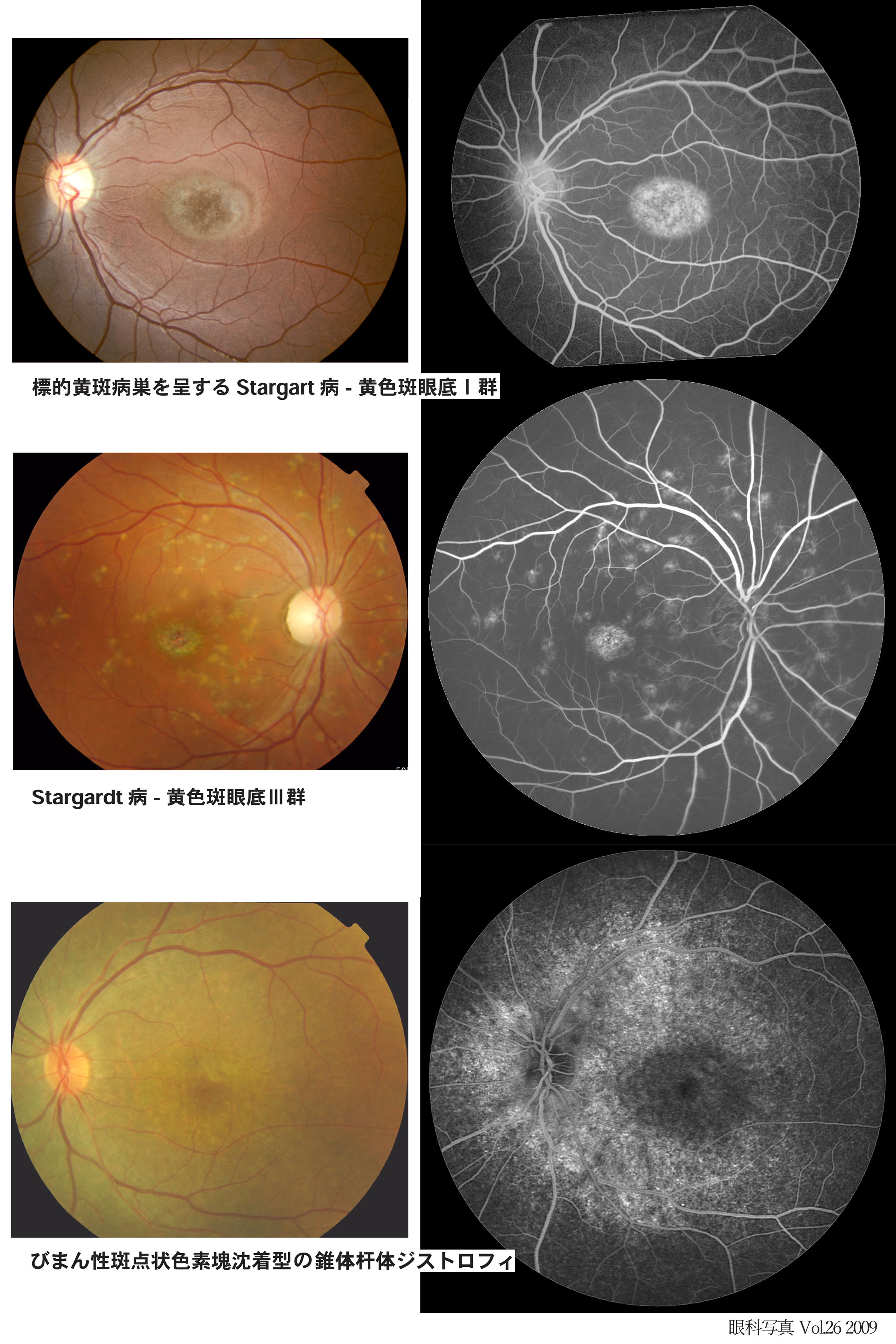
▤ subtype:➀通常型(bull's eye),➁diffuse pigment clumping 型(びまん性斑点状色素塊沈着,広範な粗糙眼底,➂脈絡膜萎縮を伴う型,が区別される(Krill).
②③は杆体障害を伴い,早期発症型として進行も早い.また②では dark choroid 所見も重要である.
▤ 錐体は網膜全体に分布し,黄斑内の錐体数は10%ほどと見積もられている.純粋に黄斑だけの錐体障害では電気生理検査に現れないだろう,とのコメントも多い.これによると,網膜ジストロフィのカテゴリーのほうが妥当,とする立場になる.
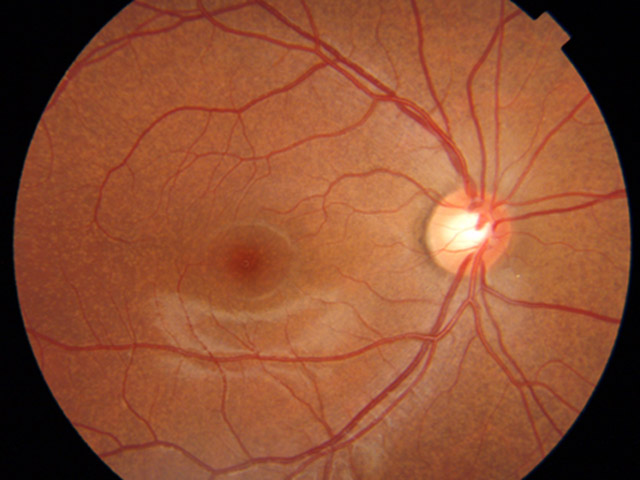
Stargardt病(黄色斑眼底群 fundus flavimaculatus)
☞ 眼底写真
黄斑を中心として視細胞が崩壊し色素上皮が変性消失する.通常小児期に両眼性の視力低下で発症する,色覚異常を伴い,若年性黄斑変性(黄斑ジストロフィ)というときの代表疾患である.眼底全体では色素上皮レベルのリポフスチン沈着の黄色斑(フレック:flecks)を認める.これにより蛍光眼底造影写真において背景蛍光が暗くなり(dark choroid,silent choroid),後極の萎縮巣は組織染を示す.自発蛍光眼底像ではフレックが強く蛍光を発し,萎縮巣は低蛍光に観察される.
進行した黄斑病巣(bull's eye)は金属粉様の反射を呈し,beatenⲻmetal (beatenⲻbronze)と形容される.なお,進行例であっても乳頭周囲では組織構造・機能が保存される,とのことである(peripapillary sparing).
一般にOCTでは網膜外層の菲薄化,特に黄斑網膜の ellipsoid zone(ISⲻOS)消失がみられる.色素上皮部あるいは網膜下に高反射隆起物(fleckに相当)を認める.進行すると神経上皮層は消失し,Bruch膜のみ残存する.
なお最初期の画像では,菲薄化に先行し外境界膜の肥厚がある.EZの消失と共にFAFを発しない年齢層や部位でみられる所見として,色素上皮細胞死に先行して視細胞の喪失が起きるという仮説を支持する.
定型Stargartd病として,ERGは準正常であることが可成りの鑑別点となる(進行度 や サブタイプ ではこの限りではない).EOG・L/D比は中等度の低下がある(黄色斑の広がりに依るらしい.診断的価値はないとのこと).暗順応の遅延もあり,進行すれば視野異常や夜盲がみられる.
| 病型 | 眼底所見 (Noble,1979 | ERG | EOG | 色覚 |
|---|
| Ⅰ群 | 黄斑萎縮巣のみ | 正常範囲 | 正常範囲 | 赤緑異常 |
| Ⅱ群 | 黄斑萎縮巣とその周囲の黄色斑(典型的なStargartd病) | 正常範囲 | ほぼ
正常範囲 | 赤緑異常 |
| Ⅲ群 | 黄斑萎縮巣と,広範に散在する黄色斑 | cone系の減弱 | 異常 | 赤緑異常 |
| Ⅳ群 | 広範に散在する黄色斑(典型的な黄色斑眼底) | cone/rod とも
減弱ないし消失 | | 赤緑異常+青黄異常 |
▤
分類の例
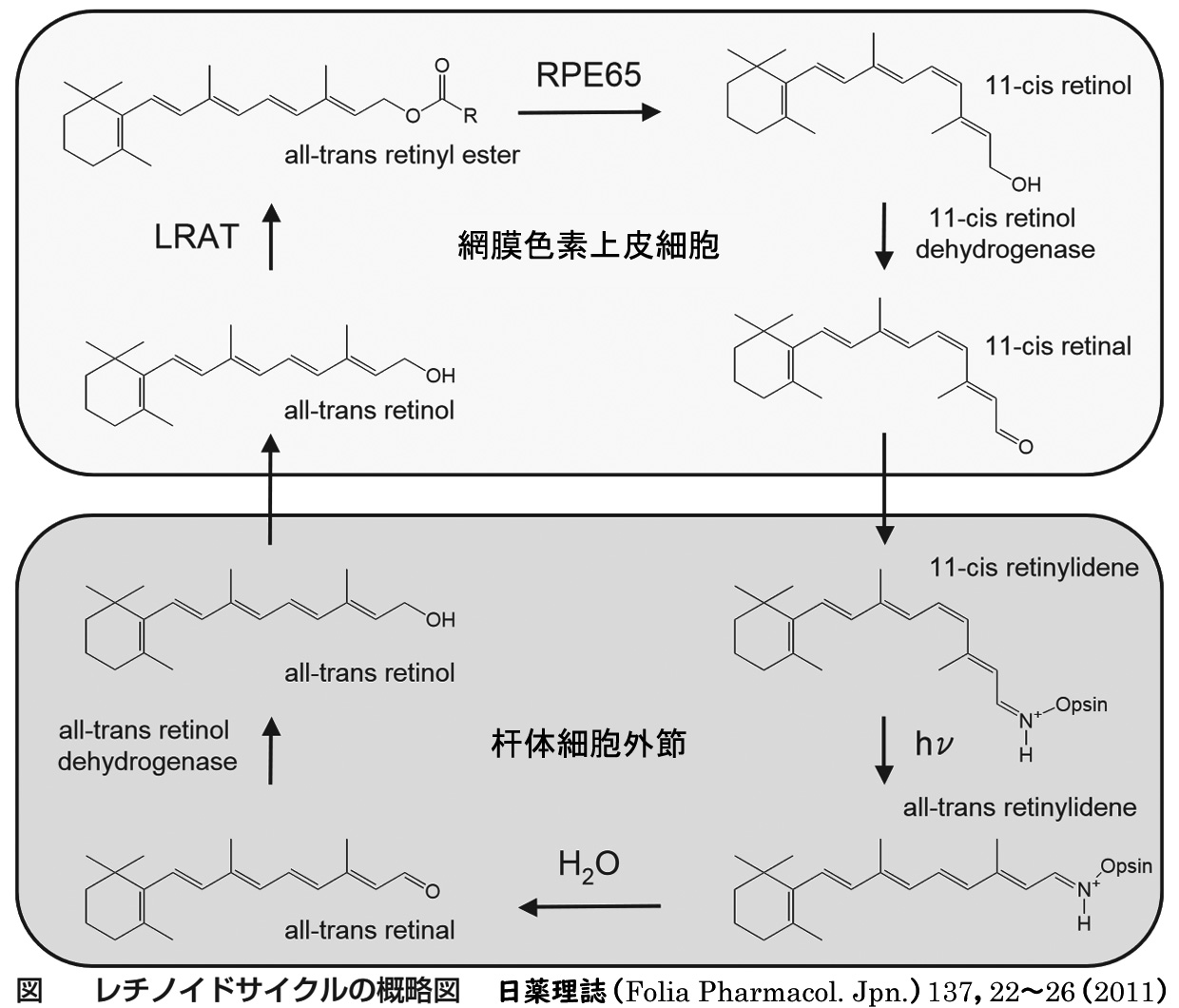
▤ 原因のひとつに,レチノール(Vit A ファミリー)の再生過程の不具合・円板膜代謝障害があり,リポフスチンの主成分となるA2E蓄積が色素上皮障害をきたす.
☞ A2Eは こちらで
▤ 責任遺伝子:
ABCA4(網膜特異的ATP-binding cassette transporter, subfamily A, member 4遺伝子;1p22):別名 STGD1(常染色体潜性(劣性)遺伝:通常型),
ELOVL4(ELOVL fatty acid elongase 4遺伝子;6q14.1):別名 STGD3(常染色体顕性(優性)遺伝:軽症型),
PROM1(4p15.32):別名 STGD4,
PRPH2(6p21.1):STGDだけでなく,多様な表現型(所見,疾患名を示す
▤ fleck 黄色斑:棍棒状,くさび状,まが玉状,小魚状
▤ dark choroid(脈絡膜背景蛍光の隠蔽現象):60~85%とのことで,認めない本症もある.黄色斑眼底の診断には蛍光眼底遺影の dark choroid所見の確認が最も重要であり,錐体ジストロフィの一部(subtype:diffuse spotty pigment clumping type)で dark choroidを呈する症例との鑑別にはERGが必須である.
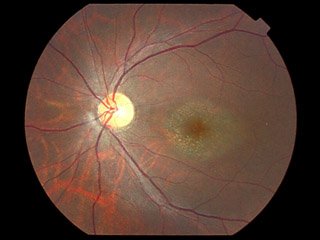
卵黄状黄斑ジストロフィ vitelliform macular dystrophy(卵黄様黄斑変性・Best病 ☞ 眼底所見は こちらで
黄斑部・網膜下に卵黄色の物質が沈着する.様相は,前卵黄(previtelliform)期 → 卵黄(vitelliform)期 → 吸収(いり卵 ∕ scramble egg)期 → 偽蓄膿(pseudoⲻhypopyon)期 → 瘢痕(萎縮(atrophic))期,の経過をたどる.症例ごとに差はあるものの卵黄様所見は思春期までに出現するとされ,視力低下を自覚する.年数と共に溶解し網膜色素上皮を含めた瘢痕萎縮の所見となる.中心窩において視細胞外節が温存されている間は,視力は良好だったりする.通常,両眼性であるが片眼性の症例もある.ある時期の断片的な所見では診断が難しかったりする.
組織学的にはリポフスチン(視細胞外節由来)様物質の色素上皮細胞内沈着は眼底広範囲に及ぶことがわかっている.卵黄様沈着物質(視細胞と色素上皮との間(網膜下)だとか)には強い自発蛍光が認められる.FAでは沈着物質は蛍光ブロックを示し,萎縮部では window decect の所見となる.
全視野ERGはほぼ正常で,EOG・L/D比の異常に特徴がある.色覚障害は比較的軽度,とのことである.
責任遺伝子;BEST1(bestrophin 1遺伝子;11q12.3),別名VMD2.常染色体顕性(優性)遺伝様式.保因者でもL/D異常を示す,とのことである.
▤ bestrophinopathy:BEST1遺伝子が関与する疾患
⑴卵黄様黄斑ジストロフィのほか,⑵常染色体劣性ベストロフィノパシー(錐体‐杆体ジストロフィと重なる,とか),⑶常染色体優性硝子体網脈絡膜症,⑷成人発症卵黄様黄斑変性症,を含む.
▤ 常染色体劣性ベストロフィン症:多発する黄色沈着物を含む色素上皮の変化が黄斑部を越えて広範囲に広がることが特徴,とのことである.
▤ 成人型卵黄様黄斑ジストロフィ:正常EOG .だいたい片眼性で,加齢性病変と見做す.遺伝性はないとされるが,PRPH2遺伝子によりパターンジストロフィのカテゴリーとなる.
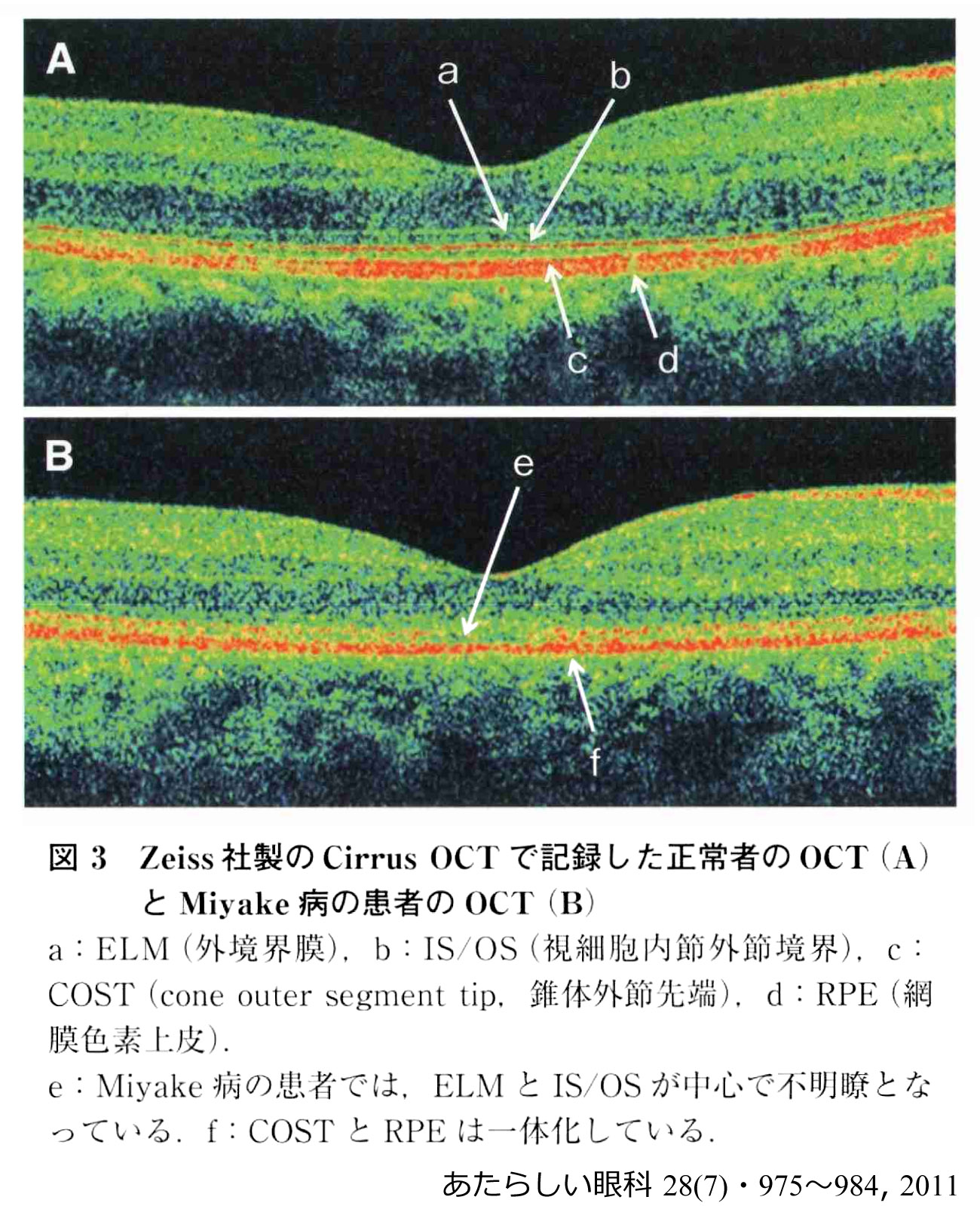
occult macular dystrophy:(潜在性黄斑ジストロフィ,三宅病
眼底所見(検眼鏡・FA・FAF)では形態変化がとらえられない進行性の視力障害.全視野ERGは正常でありながら黄斑部局所ERGのみ反応低下を示す.OCTでは視細胞外節構造の描出が不良(ellipsoid zoneの膨化・不明瞭・不連続, interdigitation zoneの消失,等)で,高齢では視細胞部(外顆粒層)の菲薄化が観察される.EZの有無,IZの有無,bulgeの有無を目印として,ステージ Ⅰ~Ⅲの分類が提唱されている.網膜色素上皮は長期保存される,とのことである.
いわゆる 黄斑ジストロフィは限局ながら錐体・杆体障害であること,錐体ジストロフィは網膜全体の錐体障害ということで,黄斑の錐体のみ選択的に障害され杆体機能は障害されにくい本症は特異的,とされる.
弱視あるいは心因性視覚障害との鑑別が重要.
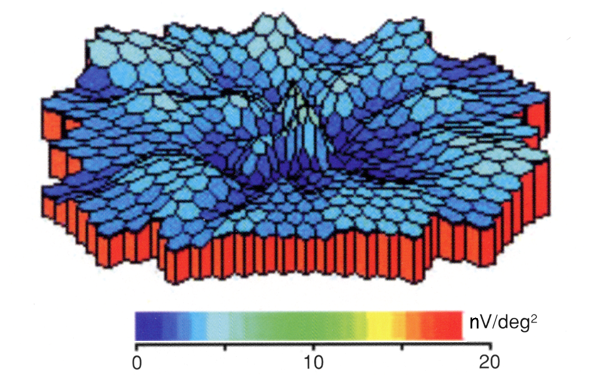
責任遺伝子;RP1L1(retinitis pigmentosa 1 like 1)遺伝子;8p 23.2 ミスセンス変異による常染色体顕性(優性)遺伝とされる.
▤ occult maculopathyオカルト黄斑症:類似する臨床所見を示しながら,遺伝学的要因の関与しない病態.
パターンジストロフィ (pattern dystrophy of the retinal pigment epithelium)
後極部での色素集積.その外観から
➀蝶形ジストロフィ (Deutman),➁網状ジストロフィ (Sjögren),➂顆粒状眼底,などと表現される.
EOG・L/D比は中等度の低下があり,色素上皮の病変を支持する.
責任遺伝子;PRPH2(peripherin 2)遺伝子;6p21.1 ,別名RDS
- クリスタリン網膜症 ☞ こちらで
網膜の後極部~広範に閃輝性小斑の結晶を生じる.結晶はRPE周囲に沈着しRPEの萎縮をきたす.続発して視細胞が変性する.
責任遺伝子;CYP4V2遺伝子による常染色体潜性(劣性)遺伝形式.